
Rare diseases can often be progressive, chronic and fatal. Approximately 72 percent of rare diseases are genetic, and around 70 percent of rare genetic diseases emerge in childhood.
Sadly, one-third of children with rare diseases die before their first birthday.
Out of over 7,000 rare diseases, only 5 percent (or less) of rare diseases are thought to have approved treatment options, known as “orphan” therapies.
However, there is reason to be hopeful. Awareness of rare diseases is growing, and with a better understanding of the pathophysiology of many rare diseases, innovative treatment options are emerging, like gene therapies that can treat the root cause of rare genetic diseases and potentially provide long-term symptom relief, or even a definitive cure.
According to Dr. Melanie Blank, clinical team leader for General Medicine Branch 1 at the US Food and Drug Administration’s (FDA) Division of Clinical Evaluation and Pharmacology/Toxicology (DCEPT), the agency is seeing one or two new applications coming in every week for new gene therapies for different diseases.
“Rare disease clinical trials are complex due to the additional scientific, medical, operational and regulatory requirements of newly emerging advanced therapies, such as gene therapy,” says Dr. Terence Eagleton, MB BS, Senior Medical Director at the global clinical research organization (CRO) Medpace. “Thankfully, we can anticipate that many more of these new treatments are expected in the future.”
Dr. Eagleton recently spoke on a webinar with his colleagues from Medpace about lessons learned from successful approaches from rare disease and gene therapy product approvals. In the same webinar, Senior Medical Director Dr. Marco Tangelder, MD, PhD, shared important medical considerations for gene therapy studies for rare diseases.
The webinar included operational strategies for these trials from Laura Omoboni, MS, Executive Director of Clinical Trial Management. In addition, Dr. Trevor Walker, DPhil, Senior Director of Regulatory Affairs, discussed the regulatory approaches.
Watch the free, on-demand webinar and gain valuable insights from this team of clinical research experts. Read on to learn about ten things that should be considered when planning a cell or gene therapy clinical trial for rare diseases.
1. The Recent Increase in Cellular and Gene Therapy Product Approvals for Rare Diseases
As of December 2022, the FDA’s Office of Tissue and Advanced Therapies (OTAT) has approved 27 cellular and gene therapy products. Last year, the FDA approved five novel cellular and gene therapy products with orphan drug designation.
This represents an increase in the number of OTAT’s novel approvals in rare diseases because OTAT approved four novel products in 2021 with orphan drug designation and only one novel product in 2020 with orphan drug designation.
The European Medicines Agency (EMA) initially recommended six novel advanced therapy medicinal products (ATMPs) with orphan designation in 2022, but the orphan designation of one of these ATMPs was later withdrawn.
This is an increase from the EMA’s approval of two novel ATMPs with orphan designation in 2021 (although the marketing authorization for one of these ATMPs was withdrawn in the European Union in November 2021) and three novel ATMPs with orphan designation in 2020.
Medpace’s work contributed to two gene therapies that gained marketing authorization approval in 2022.
2. How Can Study Protocols Be More Effective?
A gene therapy study protocol must be scientifically robust, feasible and provide clear guidance to investigators. To achieve this, Medpace works in multi-disciplinary teams of experts to jointly develop and execute advanced therapy protocols.
“Bear in mind, particularly for Phase III pivotal studies, that what you study is what you’re getting on your label,” says Dr. Tangelder. “Your protocol basically sets the scene for your label and the future use of your products in clinical practice.”
It is also vital to include the patients who can benefit the most from the gene therapy. Choosing the right patients is heavily dependent on the rare disease studied and the goal of the therapy.
Dr. Tangelder gives the example of how patients with severe hemophilia are usually included in gene therapy trials that aim to relieve them from weekly intravenous prophylaxis use to prevent bleeding. In contrast, Dr. Tangelder says that for some retinal or neurodegenerative diseases, patients who have sufficient function preserved are selected if the goal is mostly to slow or halt disease progression.
Finally, study outcome measures or endpoints need to be validated and relevant to the rare disease. For rare disease studies where there is little or no clinical trial experience, outcome assessments would have to be validated within a clinical development program.
Dr. Tangelder gives the example of the Phase III trial of the gene therapy Luxturna (voretigene neparvovec-rzyl) for retinal dystrophy due to two mutations in the gene RPE65. Natural history data and previous interventional studies were lacking at that time for this ultra-rare disease. Therefore, a multi-luminance mobility test was developed and validated as the novel primary endpoint (Figure 1), where participants had to navigate a course with varying luminance levels.

Figure 1. Novel endpoint development in the Phase III trial of the gene therapy Luxturna for retinal dystrophy due to RPE65 mutation. Reference: Chung DC, et al. Clin Exp Ophthalmol 2018; 46(3):247-259.
Although this mobility test is great for a clinical trial setting, it is useless in clinical practice since clinics cannot conduct such a large visual mobility test. To overcome this issue, the Phase III study of Luxturna demonstrated the correlation of the multi-luminance mobility test with visual field testing. The visual field test is available in most ophthalmology practices across the country and allows for easy monitoring of treatment efficacy.
3. Choosing an Appropriate Control Group
Choosing an appropriate control group is another difficult aspect of a rare disease gene therapy studies. Dr. Tangelder discusses three possible control groups for rare disease gene therapy studies (Figure 2).

Figure 2. Some options of control groups that can be used for rare disease gene therapy trials.
Using an external control group, such as a natural history population, can be challenging. The characteristics of the population and the way the outcome assessments were defined or measured in the natural history study are usually different from the clinical trial.
“A better choice is an intra-patient comparison where you measure the outcomes of interest in a lead-in phase of the study and compare that with the outcomes after administration of gene therapy,” says Dr. Tangelder.
“These are, in my view, the most valid comparisons, particularly in inherited diseases where the genotype is obviously the same in each individual patient, and as a consequence, the difference in phenotype after gene therapy is a true difference,” he adds.
Another example of an intra-patient comparison that Dr. Tangelder gives is in ophthalmology, where the treated eye can be compared with the fellow eye in the same patient.
Finally, the randomized control group may be seen as the gold standard for clinical trials and is often preferred by regulatory agencies.
However, Dr. Tangelder gives two reasons why randomized control groups are not always an option for rare disease gene therapy trials. Firstly, it may not be feasible to have sufficient sample sizes to generate comparable groups and significant statistical analysis. And secondly, patients may not be willing to take the chance to be allocated to a control group, although this can be mitigated by offering the gene therapy after a certain follow-up period.
4. Who Are the Right Patients?
Receiving a one-time, potentially life-changing gene therapy makes a big difference in the lives of rare disease patients. So, first things first — study participants and their caregivers need to be well informed about the clinical trial.
“Patients should be motivated for all study assessments, for long-term follow-up, for accepting risks and disappointment in case of lack of efficacy,” says Dr. Tangelder.
In terms of exclusion criteria, participants should have limited comorbidities, particularly in the target organ for gene therapy delivery. “For example, for liver-directed gene therapy, I recommend excluding patients with liver fibrosis or high liver enzymes at screening,” explains Dr. Tangelder.
Lastly, patient retention is important for long-term safety and efficacy documentation. Patients and investigators should recognize the importance of follow-up visits and keeping in contact regarding the trial.
5. Getting Gene Therapy Trials Off to a Good Start
A frequent challenge in rare disease clinical trials is providing clinical expertise to a small number of participants who can be dispersed all over the world.
Conducting clinical trial feasibility for rare diseases involves:
- Identifying the countries that have the rare disease population
- Finding qualified and experienced investigators and site staff
- Understanding regulations and local requirements to complete the trials
- Determining the different timelines that are needed from the beginning to the end of the trial
For the operational perspective, Omoboni explains how a good feasibility assessment involves identifying the pharmacies who are qualified and skilled in storing, preparing, administering and discarding gene therapy. This is important since not all pharmacies and sites are eligible to prepare and administer gene therapy due to biosafety and technical reasons.
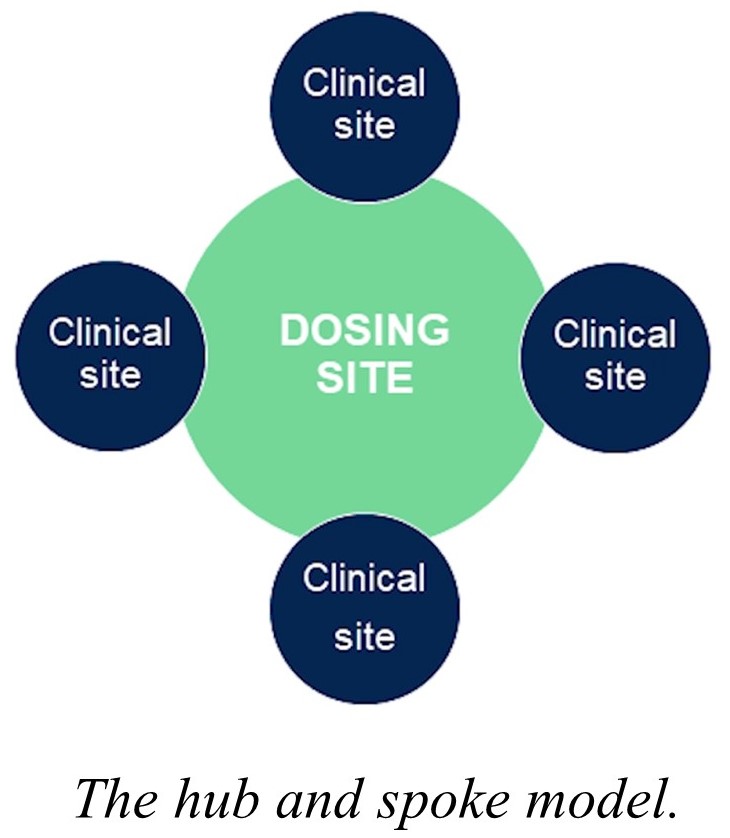
During the webinar, Omoboni discusses how the hub and spoke model is frequently applied in rare disease gene therapy studies. In this model, a large, central dosing site supports several other geographically dispersed clinical sites involved with patient identification, screen and follow-up. Suitable systems for data protection and data sharing between sites are required, as patients may visit a different site.
Omoboni says that patient travel is still likely required, which can be easily handled with travel support.
“We have the possibility to support patients who need to travel from region to region or even country to country,” says Omoboni. “This means organizing the travel, arranging the translations and of course having all the approvals in place for cross border patient transfer, as well as insurance/indemnity coverage.”
6. Full Confidence on the Day of Administration
It is vital to make patients feel comfortable and at ease, particularly on the day of administration. Patients know very well that they are receiving a one-time treatment that may be life changing.
There needs to be good communication between sites if patients are referred for the gene therapy administration. Omoboni also stresses the importance of detailed planning and preparation for administration day.
“There needs to be a mock run done with sites and pharmacies. This also helps ensure that there is a good communication pathway and transfer process between the pharmacy and the clinical team,” she says. “The sponsor and CRO medical and operational teams also need to be fully available for immediate site support.”
7. Avoiding Issues in Long-Term Follow-Up
FDA guidelines suggest that studies using adeno-associated viral vectors follow patients for at least five years, while for integrating vectors and genome-editing products, a minimum 15-year follow-up period is recommended.
What are the best ways to engage patients for long-term follow-up visits?
“Medpace did experience that you can follow-up with patients during their life changes and avoid losing any data from the long-term follow-up phase of the study,” says Omoboni.
“If a patient is moving to another city, you can try to find another site that will be closer to them or open a new site if that is important. If the patient is moving to a country where the study is not running, you can still reach the patient and collect samples for a central lab assessment,” she adds.
In addition, educating patients and sharing the results with them can stimulate their willingness to comply with long-term follow-up visits and assessments.
8. Regulatory Readiness
Rare disease trials often need to take a global, multi-center approach to study design to achieve recruitment targets.
“Conducting global studies, especially at an early phase, provides global clinical data that can potentially be used to support approvals across multiple regions,” says Dr. Walker. “Sponsors should be thinking about a global regulatory strategy in advance and considering the regulatory environment for all of the participating countries.”
Early and ongoing engagement with regulators greatly helps clinical development planning. The use of designations, such as the Orphan Drug designation, and expedited programs like the accelerated approval pathway can help streamline development.
Scientific advice meetings with regulators help ensure the trial design and gene therapy manufacturing will meet regulatory expectations.
“Gene therapies have extensive manufacturing information that is highly technical in nature,” says Walker. “It needs to be clearly documented with as much information included as possible.”
9. Questions to Expect from Regulatory Authorities
Regulatory authorities and ethics committees or institutional review boards (IRBs) will likely have an extensive number of questions about the gene therapy clinical study.
Walker discusses how Medpace recently did an analysis of the regulatory queries they have seen for gene therapy studies, and the most common questions were about:
- Chemistry, manufacturing and controls (CMC)/quality-related questions (40%)
- Protocol-related questions (28%)
- Supporting data from clinical and non-clinical (18%)
Medpace uses their analysis of regulatory queries to help sponsors anticipate and mitigate risks when they are going through the startup phase of a gene therapy study.
10. Meeting GMO Regulatory Aspects
Despite the exciting progress, the clinical development of gene therapies for rare diseases is challenging due to additional regulatory requirements. One substantial obstacle is from the additional regulatory requirements of gene therapies containing genetically modified organisms (GMOs).
In the European Union (EU), somatic cell therapies that have undergone ex vivo gene transfer and recombinant viral vectors used for in vivo gene therapy will usually be classified as GMOs.
As a result, there are several EU regulations and directives to follow. Some EU member countries consider clinical trials with gene medicines as “contained use” according to EU Directive 2009/41/EC, while others evaluate them as “deliberate release” according to EU Directive 2001/18/EC. This is complicated even more when EU member states transpose these EU directives differently into national law.
In contrast, the US FDA has recommended that gene therapies do not need to undergo a separate GMO risk assessment in most cases. The US FDA recommends that gene therapies (and all other drugs) must meet environmental risk assessment (ERA) requirements.
How can sponsors avoid delays in study start-up when there is this lack of harmonization?
Walker says that information for gene therapies and ERA information needs to be submitted for review by biosafety committees and environmental competent authorities. “The information needs to focus on viral shedding and any risk to the environment or health care providers. Pre-submission consultation is important as it may address issues early and prevent delays,” explains Walker.
In addition to risk assessment requirements, Walker discusses how country-specific label requirements must be considered, including precautionary statements to indicate GMO contents. The shipment of gene therapy products also needs to be in line with local and international regulations.
“Granting of import licenses is required, and for gene therapies that can require additional documentation if they are classified as GMOs,” says Walker.
There are also special requirements to be considered for the shipment of biological samples for lab assessment, which may contain GMO components. If local depots are used for the shipment or storage of gene therapies, they need to be licensed for handling of GMO-containing products.
“We know that the more rare disease advanced therapy studies we do, the more we learn and the greater our expertise,” says Dr. Eagleton. “Hence, the more likely we are to succeed in delivering transformative gene therapies to patients with serious unmet medical needs in rare diseases.”
To learn more approaches for achieving cell and gene therapy approvals in rare diseases, watch Medpace’s on-demand webinar.
This article was created in collaboration with the sponsoring company and the Xtalks editorial team.





Join or login to leave a comment
JOIN LOGIN