Update (August 13, 2019): Zolgensma might be too expensive for the spinal muscular atrophy market, but should it be marketed at all? Last week, the FDA announced that Novartis submitted manipulated data in their drug application and did not inform the agency until one month after the drug’s approval. The company was quick to release a statement clarifying that data manipulation only pertained to “a specific animal testing procedure used in the development of the product” and that the assays in question are “currently not used for commercial product release”. While the FDA believes the drug should remain on the market, Zolgensma’s approval would have been delayed had the agency been aware of the misstep. Now, Novartis CEO Vas Narasimhan has to explain how the company managed to slip manipulated DNA past one of the most important regulators in North America.
Originally published on May 29, 2019:
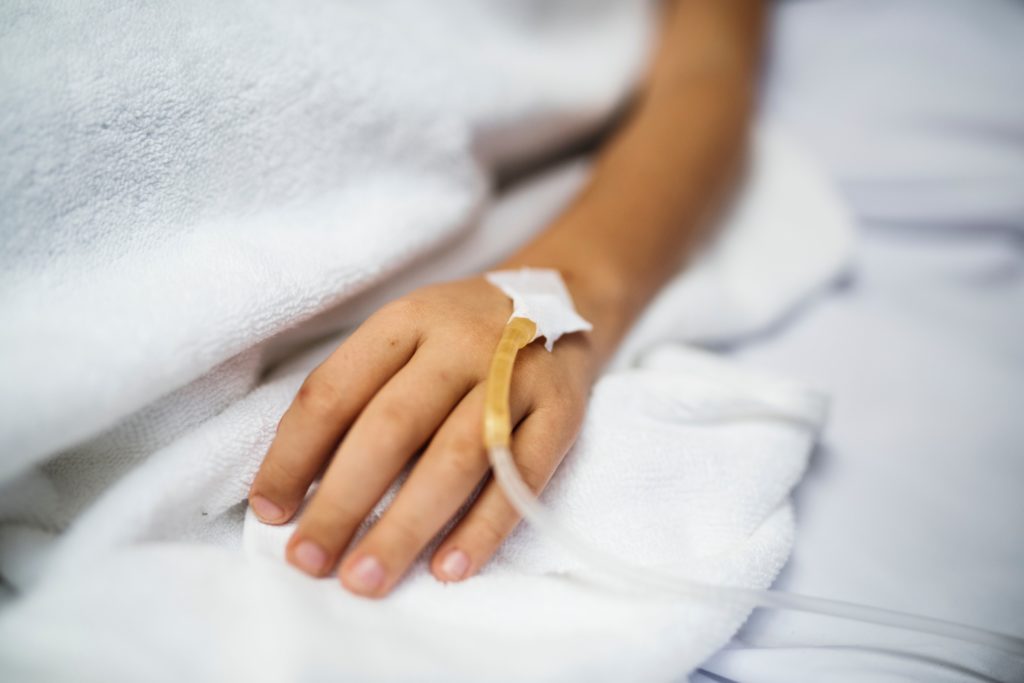
The second-ever gene therapy received US Food and Drug Administration (FDA) approval last Friday, giving hope to infants with a severe form of spinal muscular atrophy (SMA), a devastating genetic disorder that kills most infants by their second birthday. To get that one-time dose of Zolgensma (onasemnogene abeparvovec-xioi) it’s only going to cost you (or more likely, your insurer) $2 million.
According to Novartis, the new owner of the drug’s developer AveXis, $2 million is considered highly cost-effective.
“We have used value-based pricing frameworks to price Zolgensma at around [50 percent] less than multiple established benchmarks including the 10-year current cost of chronic SMA therapy,” said Vas Narasimhan, CEO of Novartis, in a press release. “In addition, the price of Zolgensma is expected to be within the range of traditional cost-effectiveness thresholds used by ICER when updated for its full labelled indications.”
It may be cost-effective in the long run, but can a young, uninsured family afford that steep price? Novartis has worked out a five-year payment plan asking for $450,000 annually, which is contingent on the drug’s effectiveness over that same time.
In SMA, a mutation in the SMN1 gene causes motor neurons in the brain to break down, interrupting the signals sent from the brain to the muscles. Because muscles aren’t receiving any signals, they become smaller or they atrophy. Children with bi-allelic mutations in the SMN1 gene usually never reach motor milestones and rapidly develop problems breathing, swallowing and sitting up on their own.
Zolgensma delivers a functional copy of the SMN1 gene that codes for a protein critical for normal motor neuron development. In theory, the motor neurons bearing the working copy of the SMN1 gene should prevent progressive muscle wasting, essentially “curing” the patient.
The clinical data leading up the approval sounds promising. In the clinical trial START, all 12 patients in the high-dose Zolgensma group were able to breathe without permanent ventilation by age 2. In the ongoing trial STRIVE, just under half of the treated patients were able to sit without support, something that untreated patients are never expected to accomplish.
However, the jury’s still out on whether children will continue to improve beyond those first few years. For one, Zolgensma calls for a one-time intravenous infusion, which may cast doubt on how effective that single treatment could really be. The only other treatment for SMA available is Biogen’s Spinraza (nusinersen), which must be administered chronically every four months and comes at a cost of about $4 million.
Zolgensma is now the most expensive drug on the market, stealing the spotlight from Luxturna (voretigene neparvovec) the gene therapy for inherited blindness, which costs a mere $850,000. Gene therapies are typically more expensive than other forms of drugs owing to the time, specialized equipment and personnel required to manufacture it.
On top of that, rare disease drug makers appear to have more pricing power when their products reach the market. Both SMA and inherited retinal disease are rare diseases, affecting only 1 in 10,000 and 1 in 200,000, respectively. According to Michelle Petersen, Senior Associate Director of Clinical Trial Management at Medpace, rare disease drugmakers need to recoup the costs of R&D to keep their pipeline going, thus justifying a high price tag.
At the same time, the FDA provides many incentives (like waived fees) to encourage pharma companies to continue rare disease research. However, some critics find this approach to rare disease research might be more harmful than helpful.
“Biopharma has been entirely redirected to rare diseases because the market will tolerate any price and the FDA will require pretty minimal data,” told Peter Back, Direct for the Center for Health Policy and Outcomes at Memorial Sloan Kettering Cancer Center to STAT News.
AveXis received Fast Track, Breakthrough Therapy, Priority Review and Orphan Drug Designations from the FDA — setting AveXis up for an expedited path to market. Roche received Priority Medicines (PRIME) status from the European Medicines Agency (EMA) last December for its own SMA drug risdiplam, and plans to seek US approval later this year.
The FDA continues to support the investigation of novel gene therapies which can change the lives of millions of people with genetic disorders.
“Today’s approval marks another milestone in the transformational power of gene and cell therapies to treat a wide range of diseases,” said Acting FDA Commissioner Dr. Ned Sharpless in a press release. “The potential for gene therapy products to change the lives of those patients who may have faced a terminal condition, or worse, death, provides hope for the future. The FDA will continue to support the progress in this field by helping to expedite the development of products for unmet medical needs through the use of review pathways designed to advance innovative, safe and effective treatment options.”






Join or login to leave a comment
JOIN LOGIN