
A container closure system consists of all the packaging components that contain and protect a pharmaceutical product. It includes both primary components, which are materials that are in direct contact with the product, as well as secondary packaging components that provide additional protection but are not in direct contact with the product.
Packaging components include the raw materials, auxiliary packaging, as well as containers and closures, and the combination of products are referred to as the system, while the product or package product refers to the substance stored in the container or system.
Container Closure Systems
A container closure system protects the product from environmental conditions, safeguarding a product’s purity, strength, safety and efficacy. The container closure system of a product is crucial for a product’s integrity, particularly for pharmaceuticals, biologics and biotechnologies that are used in treatments for patients.
Container closure systems are highly regulated by health agencies. They provide specific guidance on packaging requirements for closed closure integrity (CCI) and general integrity testing. This includes ensuring that the integrity of a product is maintained under appropriate storage conditions and that it can withstand external perturbations such as physical disturbances (i.e. shaking, changes in pressure, etc.). This involves careful selection and development of packaging systems with considerations of stability, storage, accelerated aging, freeze-thaw, altitude, drop, vibration distribution simulation, as well as identification and characterization of compounds released from packaging materials (i.e. extractables and leachables).
Manufacturers must conduct various tests such as stability and extractables and leachables (E&L) testing for packaging materials and enclosures to meet safety and efficacy guidelines.
In the US, the Food and Drug Administration (FDA) and the United States Pharmacopeia (USP) offer guidance for the development, application and use of appropriate container closure systems through the integration of analytical tests that ensure product integrity and compatibility.
In a webinar presented by Curia (formerly AMRI), experts from the company discussed primary packaging solutions for pharmaceuticals and medical devices, including the different types of tests required by regulatory agencies to ensure that a product’s container closure system is adequate.
Curia is a global contract research, development and manufacturing organization (CDMO) that specializes in drug discovery, spanning the entire product lifecycle from target to lead candidate identification, development, manufacturing feasibility, through to commercialization and post-marketing. With a network including North America, Europe and Asia, Curia aims to improve patient outcomes and quality of life by offering a complete suite of solutions across its three divisions: Discovery, Development and Analytical (DDA), Drug Product and API Manufacturing.
In the presentation, some of the testing strategies used to select and qualify packaging components and their use together as packaging systems were discussed.
Requirements for Primary Packaging Components
USP and EP Organizations for Container Regulation
Containers undergo rigorous qualification based on their material of construction and intended use. The USP and the European Pharmacopoaia (EP) have minimum requirements for a container based on these parameters.
Recently, both the USP and EP made updates to their chapters. While USP introduced chapters 661.1 and 661.2, which have better-defined parameters for material and system requirements for containers and container closures, EP introduced more stringent autoclave parameters (such as defined time cycles, temperature and pressure) for extraction conditions. Given that these parameters are difficult for most autoclaves to achieve, some issues with supply have arisen as laboratories and producers strive to conform to them.
Charles Felter, Site Head at Curia New Jersey, LLC in Lebanon, NJ explains that the new USP chapters often require more testing than the original Chapter 661, which is part of the reason that USP granted an extension (until 2025) for full adoption in collaboration with external stakeholders.
Parameters and Tests
Functional tests are conducted to ultimately ensure patient safety, which is the end goal of any container closure system. For example, protection against changes in moisture content (gain or loss) is an important container performance consideration for oral liquid and solid doses, which is evaluated through permeation or moisture transmission testing. Closures and ancillary packaging must also be assessed chemically to ensure performance and purity.
Internal packaging materials, such as cotton coil and rayon, desiccant packets or canisters and their contents, also require verification. Other tests may include biological reactivity, elemental impurities and identification, depending on the material of construction and intended dosage form.
Other tests include dose accuracy qualifications for containers purposed for dose measurements measure, while lot consistency is ensured through dimensional tests. Ensuring a properly-sealed final product involves several measurements and various thresholds, depending on the equipment used for filling and finishing, as well as the tolerances for fit.
The International Standardization Organization (ISO) also provides guidance to ensure that needles, syringes, Luer locks and auto-injectors function within acceptable limits.
Stability Studies
Stability, cycling and distribution simulation studies are conducted to evaluate the impact of environmental conditions on complete packaging systems, and in turn, product quality, that a product may experience over its useful lifespan. Environmental conditions can affect product shelf life and the viability of product formulations.
A stability program involves challenging a packaging system to corroborate shelf life label claims, depending on the packaging components used. Depending on the nature of the product, various conditions and timings are employed, explains Felter. Considerations include the type of product and packaging system, as well as labeled storage conditions.
Typical conditions for stability programs involve testing different temperature and moisture conditions. For example, for biologics and cold-chain storage products, products are evaluated at temperatures of -80°C, -20°C and 2 – 8°C.
Felter says products that undergo temperature changes should undergo freeze-thaw cycling studies to ensure these changes and conditions don’t degrade the product or compromise the packaging system.
Packaging Testing
There are several tests for container closure components, the container closure system and secondary components related to logistics and transport that will ensure that the correct container closure components have been selected and are adequate to ensure the quality of the end product.
Secondary and tertiary packaging systems must also be qualified when loaded with the primary system. This includes simulated distribution testing, force testing and label testing. Some common standards are controlled by ASTM (American Society for Testing and Materials) and ISTA (International Safe Transit Association).
Some of the different types of tests include:
Vibration testing: simulates the vibration that occurs during various shipment methods of the packaged product, including by truck or plane.
Drop testing: ensures the integrity of the packaging system through controlled drops or shocks, which may happen through the course of storage, shipment and delivery. Drops can be performed on shippers, metal cases, drums and cartons. Some specialized examples include determining the most protective cassette design for cryogenically frozen pouches or the maximum height a product can be dropped at before damage to it is incurred.
Compression testing: simulates stacking of boxes for shipment or warehouse storage. This can include the load of a full pallet and may increase up to a measured failure point of the package system. Performance of the package and packaged product under various pressure conditions including at altitude can be evaluated i.e. a programmable low pressure chamber can be used to simulate air pressure on an airplane up to 40,000 feet.
Gas exchange is another important consideration with respect to changes in pressure and temperature. In addition, various leak-detection technologies, including altitude chambers, can be used for some probabilistic leak detection methods (such as bubble leak and dye leak) to detect leaks in the packaging system.
Aaron Liss, Business Development at Curia, says that CCI testing (CCIT) is “essentially an elaborate term for leak testing performed in a very sensitive and quantitative fashion.”
Packaging Systems – Integrity and Compatibility
According to Liss, the best practice is to integrate simulated distribution and stability studies with CCI. Therefore, once the shipping qualification cycle is completed on a pallet of product, those same units should be utilized for CCIT validation.
CCIT
CCIT involves packaging integrity testing and leak testing. Previously, guidance was limited on CCIT, despite the concept of assessing the integrity of sterile pharmaceutical closures and sterile implantable medical devices having been around for decades, explains Liss.
Currently, there are guidance documents from the FDA, PDA, USP and the EU Annex I (which is currently in draft form and under review).
USP <1207> provides guidance on the integrity assurance of product packages (sterile products).
A significant change to USP <1207> was the elimination of microbiology testing due to the introduction of more favorable methods. The current guidance chapter defines the concept of CCI more broadly (going beyond just traditional microbial challenge testing) to encompass the absence of any kind of package leaks that risk product quality.
The revised definition is therefore more ‘leak-oriented’ – a package is deemed to have integrity if it allows no greater leakage than the product’s maximum allowable leakage limit – or in other words, the absence of the largest and smallest leaks of concern.
The FDA guidance document highlights a relatively recent industry trend (seen in the last five years) of companies replacing sterility testing during stability with a deterministic CCI method.
The European counterpart – EU Annex I – has also undergone changes, including a major focus on CCI and supportive statistics based on data collected using deterministic methods.
Package Integrity Testing
Leak testing is distinct from permeation, and thus the two should not be confused, says Liss. Permeation occurs when a small fraction of molecules are able to move through a barrier by way of any one hole. A non-porous package is able to permit permeation, but not the volumetric flow of air, which CCI evaluates.
Some common definitions for integrity testing in USP <1207> include:
Inherent Package Integrity: a measure of the leak tightness of a container-closure system, given anticipated variables of material, composition, dimension, processing, assembly, as well as package storage, distribution and use. It involves a measure of the leak rate (or leak size) of a well-assembled container-closure system using no-defect package components.
Container Closure Integrity (CCI): ensures prevention of product loss, microorganism ingress and limits entry of harmful gases or other substances.
Leak: a breach in a package wall or a gap between package components that may permit the passage of gas, liquid or solid matter. Leak is synonymous with leak path.
Despite the rigor of each of the tests, Liss stresses that, “No one test is appropriate for all packages, or for all leak testing applications.”
However, there is common structure to any deterministic method, with all deterministic CCI methods beginning with feasibility, moving into method development, validation and ending with routine sample analysis. It is at the routine sample analysis phase you can integrate CCI with your stability pulls and lot release program, explains Liss.
There are a number of criteria to consider before selecting a method or technology. Depending on the drug product and the container closure system, the physical and functional nature of both lend itself to certain feasibility outcomes.
USP <1207.1>: Package Integrity Test Method Selection and Validation – Probabilistic vs. Deterministic Methods
The main concept highlighted in USP <1207.1> is the difference between probabilistic versus deterministic methods with respect to test method selection criteria.
Probabilistic methods rely on a series of sequential or simultaneous events, each associated with random outcomes described by probability distribution.
Probabilistic leak technologies include bubble emission, microbial challenge (which has been phased out, as mentioned earlier), tracer liquid (dye) and tracer gas (helium; in sniffer mode).
Deterministic methods involve leakage detection or measurements based on phenomena that follow a predictable chain of events.
Deterministic leak technologies include high voltage leak detection (HVLD), laser based headspace analysis (HSA), vacuum decay, tracer gas (helium) in vacuum mode and airborne ultrasound/seal scan.
Below is a comparison between deterministic and probabilistic methods with respect to various parameters:
- Quantitative vs. qualitative: Deterministic methods are more objective compared to probabilistic methods which are much more subjective. Deterministic measures of leak detection are based on physiochemical technologies that are readily controlled, monitored and yield quantitative data. For example, in a high-voltage leak detection (HVLD) test, measured data is quantifiable i.e. a flux or a peak in electrical current signals a leak. However, in a probabilistic method such as a dye ingress method, an analyst may claim they see blue dye whereas another may not.
- Specificity: Deterministic method parameters are tailored and optimized to a specific container closure system and drug product, while there is little ability to modify or optimize a probabilistic method.
- Sample size: Because probabilistic methods yield results associated with uncertainties, this necessitates large sample sizes and rigorous test condition controls to obtain meaningful results.
- Detection limit: Probabilistic methods have a high limit of detection whereas deterministic methods have a low limit of detection, making the latter more sensitive. For example, the detection of an ingress method may be 20 microns. However, detection limits for High Voltage Leak Detection (HVLD) is around three to five microns, which is approximately a four times improvement in sensitivity. This is the major reason why regulatory has started rejecting ingress methods to satisfy USP <1207>.
Liss says because they are “the safety watchdogs for industry, it’s no surprise [that regulatory agencies] would gravitate towards an approach that will protect patients with higher confidence and certainty.”
While preference has shifted towards deterministic leak methods to satisfy USP <1207>, probabilistic methods still have utility such as in early R&D for clinical studies, or during qualification for simulated distribution studies for shipping.
USP <1207.2> Package Integrity Leak Test Technologies
There are various technologies for CCI deterministic testing. Technologies mainly involve the use of either tracer gas or vacuum-based modalities.
Helium Method
Helium is the most sensitive deterministic method, offering many benefits. It involves flooding samples with helium tracer gas and placing them into an evacuation chamber or fixture. A helium mass spectrometer then quantitatively measures the tracer gas leakage rate.
This method can detect leaks below one micron (around the magnitude of 0.1 to 0.3) and can be correlated to microbial ingress defect sizes. It is the only reliable instrument to detect maximum allowable leakage limits. The technology can also be used in conjunction with residual seal force for torque and capping studies.
Helium is ideal for empty, non-porous, rigid, flexible packages (blisters, vials, etc.). It evaluates performances of mated packaging components, and is best used as a design tool to demonstrate the inherent integrity of a package. It can also be used in conjunction with residual seal force (RSF) for Torque and capping studies.
Helium is the most reliable and accurate when evaluating the inherent integrity of a package system without any drug product contained inside it. It allows for the verification of the main features of the component, cap to bottle, stopper to vial, explains Liss.
He says an excellent way to leverage helium is when trying to validate a stopper-vial system for an entire product line. For instance, if there are three vial sizes and various combinations of ten to 12 different drug products are going to be put inside of them, helium is a great way to confirm the integrity before testing specific drug products contained within each closure.
Non-Destructive High Voltage Leak Detection (HVLD)
HVLD is also known as the ‘spark test.’ It is an approach for detecting the presence and location of leaks in the wall of non-porous packaging, rigid or flexible systems containing liquid drug product.
The principle of high voltage leak detection is based on quantitative electrical conductance measurements. The presence of a leak in the proximity of a conductive liquid results in a drop in electrical resistance, as evidenced by a spike in current above a predetermined pass/fail limit. This highly sensitive method even works to detect package defects clogged by product formulation proteins or salts, explains Liss.
Stability studies have supported the use of this technology for non-destructive leak testing for a variety of product formulations. However, it should be considered on a product-by-product basis, as certain drug formulations contain combustible solvents.
Package systems that may be tested by HVLD include parenteral vials, pre-filled syringes and cartridges, plastic containers and plastic bags or pouches. Auto-injectors are not ideal to test through HVLD as it involves disassembly of the device to evaluate the syringe or cartridge contained within. Acceptable package system materials include glass, plastic, poly laminate and CZ resin.
While the package system should contain a conductive solution, advances have been made to the instrumentation to also allow assessment of more non-polar liquid solutions.
The HVLD process involves several steps:
- Feasibility: HVLD uses specific fixtures for the package system and so the appropriate instrument parts must be determined.
- Method Development: the method development phase is the most laborious, and evaluates several parameters using the fixtures from the feasibility report. A total of six parameters (voltage, rotation, motion profile, speed, sensitivity, pitch) must be optimized. Each of the parameters are dependent on one another, such as they influence test results, while taking into consideration the material of the container closure system, as well as any electrical influences from the unique product properties.
- Validation: after method development, validation involves determining the characteristics of the method, and proving that results are repeatable and reproducible.
- Sample Analysis: once a validated method is chosen, sample analysis can be carried out using specific protocols. This includes beginning sterility testing and performing GMP/CCI methods during stability evaluations for release testing.
Vacuum Decay
Vacuum decay is a non-destructive test that facilitates the identification of leaks that may not be visibly detectable. It involves placing a test package into a custom-designed chamber that is then exposed to a vacuum. Sensitive pressure transducers monitor changes in chamber pressure due to package headspace being withdrawn as a result of any leaks that may be present. Using acceptance criteria established through the method development phase, quantitative test results can be judged as pass or fail.
Liss says that in their experience at Curia, vacuum decay is the most versatile technology platform for CCI, which can accommodate a broad range of drug products and package systems due to the uniqueness of various chambers that can be fabricated and outfitted with controlled vacuum evacuation.
Vacuum decay leak testing is applicable to any package containing headspace, including, but not limited to, parenteral vial packages, screw-cap bottles, auto-injectors, flexible bags and pouches. Acceptable materials include glass, plastic, poly laminate and CZ resin, while package contents that are granular, liquid (non-proteinaceous), lyo-filled and solids are permissible.
A big trend seen in the industry now is the FDA asking for deterministic CCI testing for intermediate storage containers and flexible bags for biologic products. Typically, these package systems are quite large in size, which makes vacuum ideal due to the unique and robust chamber designs available, explains Liss. As with any technology, there are some challenges to consider. For example, vacuum decay is generally not recommended for highly viscous products that have the potential to clog and mask leaks.
A vacuum decay program follows the same high-level phases as an HVLD program (feasibility, method development, validation and routine sample analysis). Whereas the HVLD uses fixtures, vacuum employs the use of a chamber to pull a vacuum.
Shown in Figure 1 are two of the latest types of vacuum decay chambers. On the left, the drawer chamber can accommodate any flexible IV bag or pouch system. Previous chamber technology was limited in size. The automated drawer in the newer version allows for any type of flexible package system to be tested; if it fits, it can be evaluated.

On the right, is a system termed the ‘Russian Doll Chamber.’ It is a stackable chamber that can evaluate several vials simultaneously. The old method involved testing each vial within the insert individually. Hence the new chamber can save on costs and resources.
Non-Destructive Laser Headspace Analyzer
Assessment of package headspace via laser analysis provides a quantitative, non-destructive measure of oxygen, carbon dioxide, water vapor or internal pressure in a non-porous rigid or non-rigid headspace region.
This technique involves use of a near-infrared diode laser to pass light through the gas headspace region of a sealed package. Light absorption, measured using frequency-modulated spectroscopy, indicates gas concentration and pressure. Headspace analysis is non-destructive and as such, it may be repeatedly performed over time on the same vial or system to provide a quantitative measure of the sample’s leak rate, which can be correlated to stability.
The technology is particularly appropriate for verifying the integrity of packages that must maintain a specific gas headspace content due to oxygen or moisture sensitivity. The technology can be used to evaluate leakage that may occur during ultra-cold or cryogenic storage, and as such, storage conditions are below the glass transition temperature of packaging components.
Examples of ideal types of container closures to evaluate with headspace include syringes, vials, cartridges and tubes. For materials, while both translucent glass (clear or amber) and translucent plastic are permissible for laser headspace, Liss says that in their experience, glass is easier to handle than plastics because plastics require careful fabrication of positive controls using conical laser drilled effects to prevent clogging and false negatives.
Laser headspace follows the same high-level stages, starting with feasibility through to sample analysis, as vacuum decay and HVLD. However, laser headspace does not involve a chamber or fixture but requires that a sample analysis kit be created with appropriate components.
USP <1207.3> : Seal Quality
USP <1207.3> is the last subsection of USP <1207> and pertains to seal quality. It must be noted that these tests are not leak tests – they are functional tests that exclusively evaluate seal quality. Liss says these tests have a long historic context. Methods include airborne ultrasound scanning, package seal strength and package burst testing, which are best suited for package designs such as Tyvek trays with lids, or Tyvek pouches. Other methods such as closure application, torque removal and residual seal force are best suited for vial/stopper configurations.
Package Compatibility
Package compatibility is another important area of testing as pertaining to extractable and leachable (E&L) compounds.
Extractables are inorganic or organic chemical entities that could potentially be released from packaging materials into a drug product under laboratory conditions. Laboratory conditions involve using model solvents with a range of polarity under ‘worst-case’ temperature and time conditions to pull out elements of toxic concern.
Leachables are inorganic or organic chemical entities that leach into a product from packaging materials under normal storage conditions or during accelerated product stability studies i.e. simulating actual use of a product or shelf life. These compounds thus become a part of the drug matrix.
E&L testing is performed to substantiate that packaging materials will not contaminate the drug product over a product’s shelf life. As such, E&L studies are designed to evaluate the package material and drug compatibility. E&L is typically only performed once (usually during phase three) for regulatory submission.
Depending on the type of product or study scope, there are three main approaches for E&L evaluations (the approach selected depends on the purpose or application, and the FDA branch the submission falls under):
- Pharmaceutical Containers Closure Systems: involves compliance with USP <661.2>, as well as adherence to the expanded Chapters USP <1663> for extractables, and USP <1664> for leachables (under the <661.2> umbrella).
- Manufacturing (MFG) Systems/Component/Processes: can ideally involve a USP approach, or a BPOG (BioPhorum Operations Group) approach. There has been debate in the industry about whether BPOG should still be considered due to the extensive resources and costs involved. From a scientific perspective, USP <665> is a better approach and is more globally recognized.
- Devices, combo products, SUS: for a medical device or combination product, one should leverage ISO 10993, specifically, Parts 17 and 18. The FDA’s CDRH (Center for Devices and Radiological Health) branch would own the filing in this case.
The approaches have key differences in study design such as number of replicates required (i.e. ISO requires triplicate replicates for extractions whereas USP only requires duplicates) and time point differences among others.
The chart in Figure 2 is an overview that assigns risk to container closure systems under USP <1664>. Typically, all liquid products, injectables, powder products and inhalation products require E&L testing. On the other hand, although marked as low-risk, E&L is generally not required for solid oral dosage products. Solid oral dose container closures typically undergo ink or adhesive permeation studies, but they are not usually performed with specific product types for leachable evaluation.
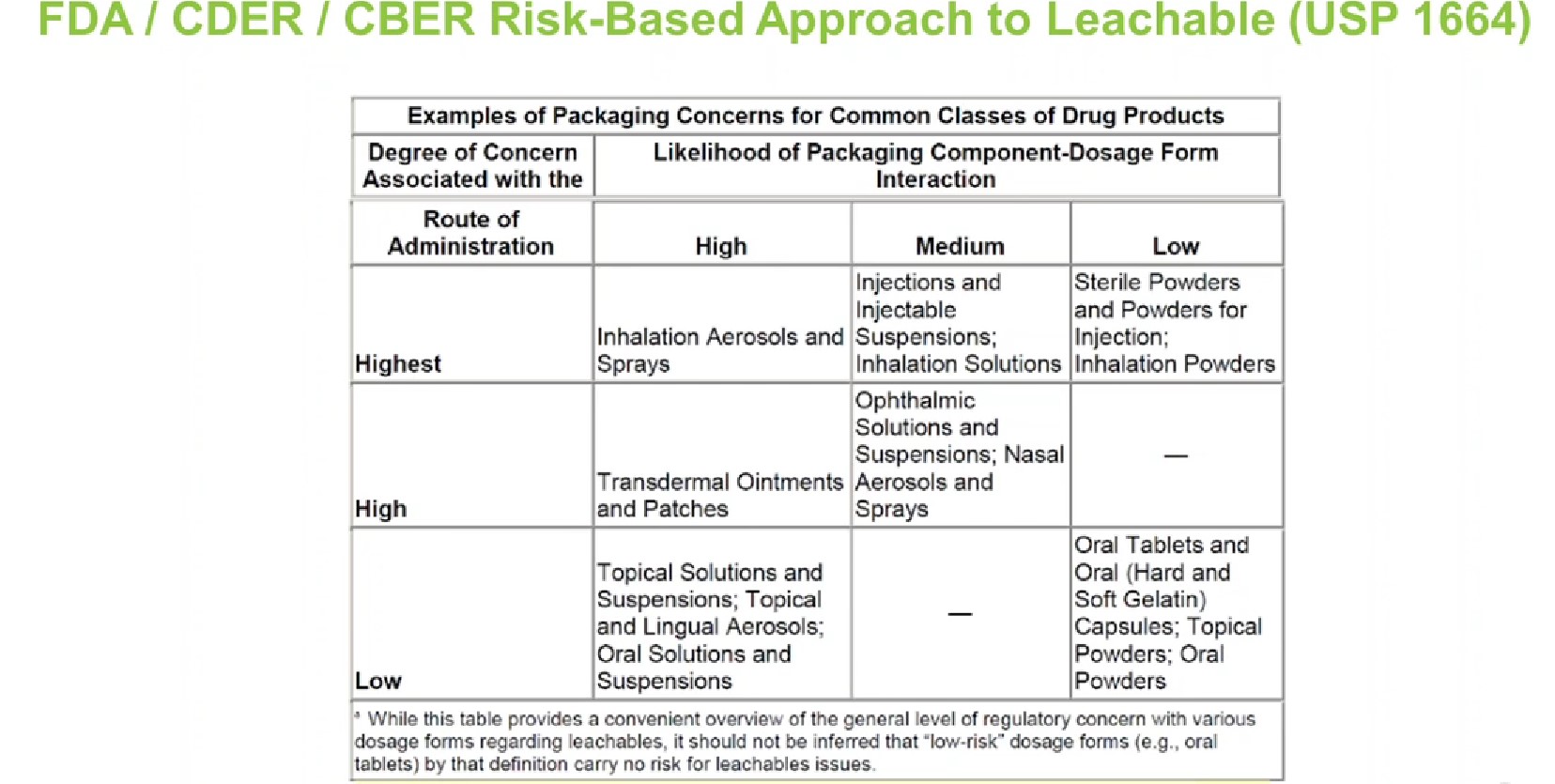
Figure 3 outlines how an E&L study program is conducted. The program begins with a controlled extraction study, which establishes the extractable profile, or chemical profile, of a pharmaceutical closure. During this phase, container closure system is challenged with model solvents with a range of polarity such as water and IPA hexane under aggressive time and temperature conditions to see what will migrate out. It is important to select appropriate solvents and conditions that will challenge package construction materials of the closure system, but that will not degrade them. For example, you don’t want to start breaking down hydrocarbons in polymeric resins, explains Liss.
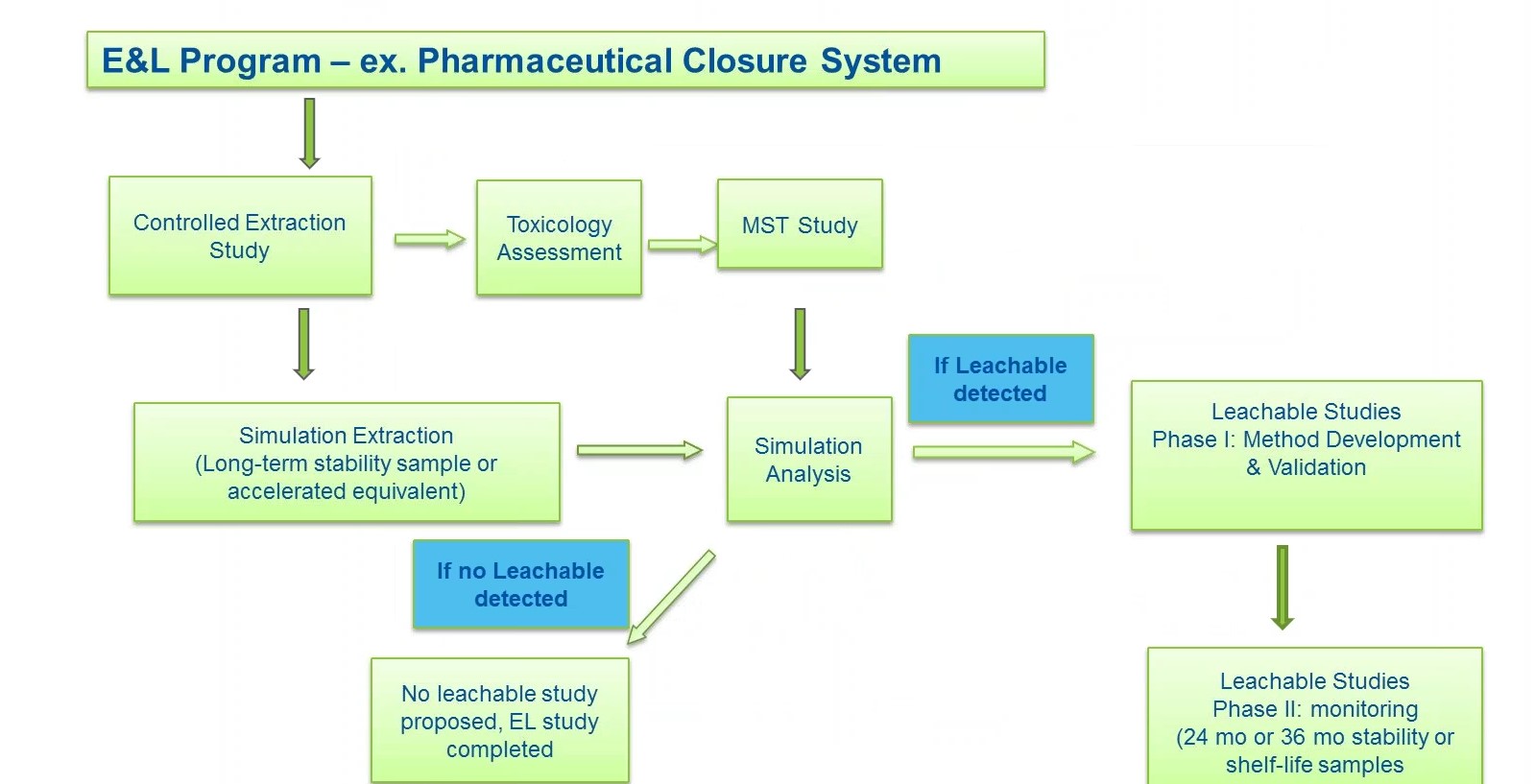
An orthogonal analysis is then performed consisting of GCMS, headspace GCMS, LCMS and ICP to generate a semi-quantitative extractable profile of all organics, semi-volatiles, volatiles and non-volatiles, as well as inorganic trace elements. Comprehensive analyses are then performed on the data to identify compounds for toxicological risk assessment (TRA). Liss says this is a critical step, as it lays the foundation for the risk assessment and the leachable analysis.
After controlled extraction, a TRA is conducted on identified extractable compounds. Compound structures are evaluated according to QSAR software to prioritize compounds from manual evaluation and permissible daily exposure (PDE) calculations. PDE is developed for compounds using relevant toxicity information and is typically compound-specific.
Method suitability testing (MST) studies are then recommended for a list of targets based on toxicological characteristics and concentrations detected. The MST connects data from the extraction study to the leachable study by demonstrating that the instruments have the sensitivity to detect the target compounds in the presence of the drug product matrix. In MST, the drug product is spiked with the targets and analyzed by the same analytical methods, i.e. GCMS, headspace and LCMS, as the controlled extraction and subsequent leachable studies. The results provide recoveries for the target extractables, which will determine the suitability of the analytical testing methods which will then be used for the leachable study.
After the MST, a leachable simulation analysis is performed with the drug product packaged in the container closure system, and stored under an accelerated condition, or stability condition for prolonged duration (of up to 24 months or the desired shelf life) to determine the possible migration of potential leachables from the container closure to the drug product. In the study, the drug product essentially becomes the extraction solvent. The resulting extract is processed for an orthogonal analysis to generate a set of comprehensive, quantitative and semi-quantitative data for the drug product stored in the intended container closure system.
Liss explains that “Most often in our experience, chemicals of concern identified in the controlled extraction study are found at a much lower concentration, below the Analytical Evaluation Threshold (AET), or not found at all during leachables.” Therefore, most E&L studies for pharmaceutical closures follow the path on the lower left side of the chart in Figure 3.
When there is a compound identified above the AET during leachables, then one must validate a GNP method to monitor that compound throughout the product life cycle, says Liss.
Toxicology Risk Assessment (TRA)
A TRA is a high-level overview of the controlled extraction study. There are some key guidance documents that board-certified toxicologists use and refer to during their evaluation. As a recent trend, the FDA now uses ICH(M7) and ICH(Q3D) for injectables and also applies them to oral solutions. Liss says, “This can cause headaches when setting the AET, because as we know, toxicity is about dosage and route of administration.”
Another trend pertains to databases used by toxicologists to establish margin of safety. FDA prefers the use of DEREK, a knowledge-based expert system for the qualitative prediction of toxicity, and SARAH, a statistical software to identify potentially toxic chemicals for assessment of genotoxic and sensitization potential. Tox Tree and grouping by Cramer rules are starting to lose favor with the FDA.
Analytical Instrumentation
It is important to ensure that the appropriate, recommended instruments are used to conduct a successful E&L study. It is also important to have a broad range of LC instrumentation to support non-volatile organic compound (NVOC) analysis, as there isn’t an established library to easily match unknowns, explains Liss.
The modern workhorse instrument for non-volatile organic analysis is an Orbitrap instrument (Figure 4). Owing to its 140,000 high-resolution and ability to detect mass accuracy to the fourth decimal place, this instrument allows a scientist or analyst to put a structure to a compound or fragment with high confidence.

Extractables & Leachables: Challenges and Pitfalls
There are a number of challenges inherent to E&L studies. The studies are exploratory in nature, particularly during the controlled extraction phase where potentially unique and novel compounds may be identified. As such unknown compound ID is a common problem.
Unknown compound identification. Unknown compound ID is “one of the primary hot buttons with regulatory agencies,” says Liss. Simply put, you cannot have unknowns show up in your extractable profile. If they do, the FDA will assume the worst-case scenario and deem the compound or fragment to be genotoxic or eugenic. There are different types of unknowns, such as compounds that are not identified correctly, or compounds that are not identified at all.
Unknowns are often identified during NVOC via LC methods. Having a robust database and library is the easiest way to overcome and deal with unknowns, but it is not always feasible. Equipment sensitivity and ensuring that the method suitability study is designed correctly to target the appropriate compounds for further leachable analysis can be other approaches to take. If all this fails, then analyst expertise and published literature research are the only other ways to help decipher the most likely candidate ID for the compound.
Product/Material degradation: It is crucial to know and understand the entire suite of materials that may come in contact with the drug product and/or patient. For example, if a pre-filled syringe uses a silicone plunger, hexane should be avoided, or if a drug product is oil-based, then a highly polar water extraction is not needed. Therefore, solvent selection is very important in extraction and should be those that challenge, but do not degrade the product or packaging materials.
Sample preparation under appropriate pH conditions is important. For example, for pharmaceutical closures, the water extract should be pH’d appropriately to mimic the drug product. With respect to physical elements, proper agitation/inversion/sonication tests are used. For example, pharmaceutical closures should be incubated and inverted so that the extract comes into contact with all material surfaces. For an implantable medical device, such as a cardiac implant, it should be fully submerged and sonicated during the extraction incubation.
Setting the proper AET: It is advisable to be conservative and utilize M7’s 1.5 micrograms per day for potentially genotoxic impurities, even when the duration of treatment is shorter than ten years, says Liss. Also correlating this to ICH(Q3A), Q3B for safety qualification and identifying the PDE based on a margin of safety. In addition, utilizing TQR best practice of safety concern threshold (SCT) of 0.15 micrograms per day, and optimizing analytical methods toward the sensitivity will also reduce questions from the agency.
Dosage and exposure are especially critical with respect to toxicity, which is a primary concern of the FDA. Thus, it is important to understand the intended patient population, route of administration and duration of use to establish a proper AET. Setting an appropriate AET, and being able to demonstrate it through the appropriate calculations, will help reduce agency questioning and avoid any retesting, says Liss.
Primary package selection should be considered early in development during drug formulation or device design. The steps and processes involved should be carried out in a step-wise approach. Individual components and materials should first be qualified using the various USP tests, after which a final container closure can be qualified. Once a design is finalized, physical and functional testing and stability and simulated distribution studies can be run simultaneously and integrated into one program. Finally, CCI and E&L testing are conducted in later stages.
To learn more about packaging solutions for pharmaceuticals and medical devices, watch this free webinar by industry experts from Curia.
In the end, the goal is to satisfy regulatory requirements for packaging materials and closures to ensure that a safe and effective product is delivered to patients.
This article was created in collaboration with the sponsoring company and the Xtalks editorial team.








Join or login to leave a comment
JOIN LOGIN