
Accelerated drug development programs and pathways help expedite the testing of drugs and biologics for rare and orphan diseases. These programs assist in addressing serious unmet medical needs in a timely manner by providing pathways for expedited drug development and approval. Given the unique patient populations and timeline considerations associated with rare disease clinical trials, it is important to thoroughly understand the regulatory guidelines that govern orphan and rare disease drug development, which can differ across regions around the world.
In an informative webinar presented by regulatory affairs experts from Medpace, some of the key regulatory strategies and considerations involved in rare and orphan disease drug development were discussed in depth.
The presentation highlighted the various designations and regulatory pathways that can be used to expedite and accelerate treatments for rare diseases in the US and European Union (EU). It outlined specific requirements for these designations, including suggestions for timely submissions and what to expect in meetings with regulatory agencies such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in the EU. Additionally, requirements associated with pediatric rare disease drug development were also discussed.
Global Acceleration of Drug Development in Rare and Orphan Disease
Obtaining accelerated approval for new drugs and biologics in the rare and orphan disease space is essential in delivering new therapies to patients with debilitating rare diseases and conditions. To facilitate this, there are expedited pathways and designations that can be utilized to engage regulatory agencies around the world both early on and throughout clinical trials and development processes.
Time is of the essence for rare disease patient populations and thus these accelerated channels can be used to gain conditional approvals to achieve earlier time to development in order to get new therapies to patients faster. Key trial measures can be achieved through orphan designations both in the US and in the EU through specific expedited pathway designations.
Christina Vonderhaar, Senior Director, Regulatory Affairs at Medpace discussed the importance of having a global mindset when considering drug development considerations as investigators should ensure that they understand the implications of receiving scientific advice from each of the regulatory agencies in the regions that they are seeking approval in. In the US, this would be the FDA and in the EU, the EMA or other national competent authorities.
Accelerated Pathways in the US
In order to understand the expedited and accelerated pathways that exist in the US for the development of treatments for serious, life-threatening rare or orphan diseases, it is first important to understand the definitions. In the US, a serious medical condition or disease is defined as being associated with morbidity and that has significant impact on day-to-day functioning.
A clear rationale must also be provided that explains why you’re looking to treat a potentially serious condition and what the serious unmet needs are; this involves being updated on the most recent literature on the condition and existing treatment paradigms, including both approved and non-approved drugs, so that this information can help guide your rationale and be clearly communicated to regulators.
According to Vonderhaar, it is especially important to “highlight potential areas where the new therapy may be more efficacious or have a better safety profile in demonstrating superiority compared to an available therapy.” Vonderhaar added that this is why, “you have to work within the larger context of the competitive landscape and not just understand your own product.” Moreover, a given patient population may not have any available therapies for the given condition, which is important to highlight in explaining how you plan to address and fill in the gap.
Any clinical development plan must therefore clearly outline and support the intent to treat a given rare disease population if sponsors are looking to obtain orphan designation or a fast track designation for a treatment.
The US FDA has three different expedited programs: Fast Track, Breakthrough Therapy and Accelerated Approval (Figure 1).

Fast Track
The fast track program involves actions to expedite development and review, including rolling reviews. One of the key criteria for fast track is the ability to address an unmet medical need in a serious condition. An important feature of the fast track program is that applications typically require the least amount of clinical data, or none at all, to support the designation. Because of this, Vonderhaar explained that oftentimes this designation or program can be the first one to embark on because it can be requested the earliest in development, owing to the minimal data required.
Breakthrough Therapy
There are several advantages to the breakthrough therapy program, which include intensive guidance on efficient drug development, organizational commitment from the FDA, rolling review and other actions to expedite review. Breakthrough therapy is a key designation for a lot of different indications particularly in oncology as well as in rare and orphan spaces because some products can be eligible for shorter meeting windows with the FDA, which can reduce the time it takes to receive guidance and feedback from the agency.
In contrast to fast track programs, some clinical data is required to demonstrate product efficacy for the breakthrough therapy program and therefore application for this designation tends to occur later in development.
Accelerated Approval
This type of approval is based on a surrogate or intermediate endpoint that can potentially predict clinical benefit. This is a common pathway for oncology indications but can also be utilized in other therapeutic areas.
Other sets of designations are more specific to particular therapeutic areas or indications. These include:
Qualified Infectious Disease Product (QIDP): This includes fast track designation and a 5-year exclusivity extension for certain applications.
Limited Population Pathway for Antibacterial and Antifungal Drugs: This application review takes into account the severity, rarity or prevalence of the infection that the drug is intended to treat. This could include a very small or a rare population within a broader indication, or a more severe population within a broader indication.
Regenerative Medicine Advanced Therapy: This includes all features of the breakthrough therapy program and involves greater interaction with agencies, which is particularly important for advanced therapies like gene therapies as there is a fair amount of new guidance for it. Qualification requires some clinical data in order to achieve designation.
Priority Review: This is a shorter review of marketing applications i.e. six months compared to the standard 10 – 12 month review.
US Rare Disease Vouchers
The requirements for vouchers for rare diseases is that at least 50 percent of the affected population is between 0 and 18 years of age where the prevalence is lower than 200,000 overall at the time of marketing approval for the pediatric indication. A voucher for priority review is issued and vouchers are transferrable.
The agency will generally require that you are not seeking an adult indication at the same time as the pediatric application is being submitted for review, although there can be exceptions to this.
With respect to timelines, disease vouchers currently must either be utilized, be applied for or have received designation by September 30, 2020 in order to have an application considered to be viable for this voucher. This date can be subject to change based on additional legislation.
Rare tropical disease voucher
This type of voucher is similar to the rare pediatric disease voucher however it is for tropical diseases. The FDA website currently has a list of tropical diseases that may fall under this category. At the time of marketing approval for the tropical disease indication, a voucher for Priority Review is issued.
Timelines and Engagement
Although for most of the different expedited designations and pathways there is a great degree of overlap between timelines throughout the drug development process (Figure 2), it is important to begin discussions with the FDA early to indicate intent to be on a particular pathway.
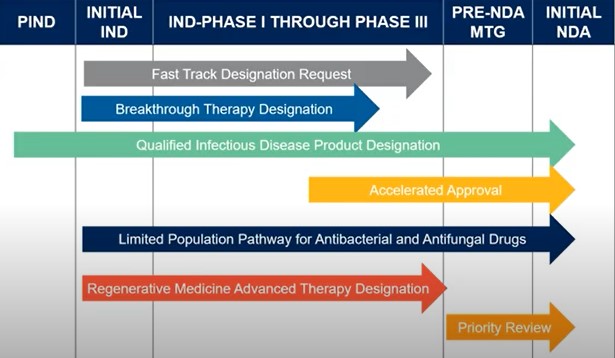
Even if there isn’t enough clinical data, it is nevertheless important to position a product in accordance to the particular designation that is being sought i.e. fast track, breakthrough, QIDP etc. Vonderhaar explained that this will allow you to obtain scientific advice at a pre-IND meeting with the FDA at which time you should begin laying out the outline for your drug as one that is intended to treat a serious condition with an unmet medical need, particularly if you’re looking at certain pathways such as a limited population pathway. She added that, “it would be important to have alignment with FDA on your clinical development plan as it may be much smaller than a typical development plan with less patients and more restrictions in terms of inclusion and exclusion criteria.”
For many of the US expedited designation requests, the agency provides a good amount of guidance pertaining to what should be included in the applications. Vonderhaar says that this “is why it’s important to review the most current guidance to ensure that you’re checking the box for each listed section so that you don’t either run into a technicality when FDA is reviewing your application or inadvertently miss a section.”
EU Regulatory Pathways
Sargon Daniel, Associate Director of Regulatory Affairs at Medpace, discussed regulatory pathways for accelerated programs in the EU. He emphasized that early engagement with the EMA is important as early conversations with the agency can help you obtain pertinent advice and guidance. An early meeting with the agency is useful in obtaining scientific advice and discussing different pathways in addition to other aspects of development including clinical and non-clinical quality.
With respect to scientific advice, a briefing pack must be produced to provide an overview of the product and development program to the EMA – this pack should be both specific and concise. Additionally, understanding the scientific advice that is given and planning with and around timelines is key.
PRIME Scheme
The PRIME scheme is aimed at improving clinical trial design and also allows eligibility for accelerated assessment at time of marketing authorization. The agency will provide scientific advice at key development milestones so that you have advice throughout the program.
Under the scheme, if a drug offers a major therapeutic advantage over an existing treatment, then the EMA will consider it to be a priority medicine.
The PRIME scheme should be discussed with the EMA very early on in any development plan. According to Daniel, “early engagement and being proactive [is necessary] because you need [the EMA] to look at your data, the robustness of your data and also to look at the risks and benefits.”
Accelerated assessment
Before submitting an application, a pre-submission meeting is required, which allows you to obtain necessary information and provide details before submission for accelerated assessment. Applicants are suggested to submit a request for a pre-submission meeting at least six to seven months prior to submission for accelerated assessment.
When applying for the PRIME scheme, this can also be discussed. Essentially, you are looking to seek guidance from the EMA, which will involve justifying the claim of your medicinal product as to why it will be needed and how it will address a serious unmet medical need.
Conditional Market Authorization (CMA)
A CMA for a treatment for a rare or orphan disease is aimed at treating, preventing and diagnosing seriously debilitating or life-threatening diseases. Applicants may be granted for such medicines where the benefit of immediate availability outweighs the risk of less comprehensive data. The goal is for unmet medical needs to be fulfilled to benefit public health. A CMA is only valid for one year, but it can be renewed annually.
Orphan Drug Development and Rare Disease: US vs. EU Regulatory Requirements
While there are many similarities between US FDA and EU EMA regulations in the area of orphan drug development, there are also some key differences. Vonderhaar emphasized that these differences are important to understand and highlight because it is imperative to “understanding your development program within the global context, particularly for rare and orphan diseases.” Therefore, an understanding of both US and EU requirements to achieve designations in both regions is critical.
The main requirement of the US FDA for orphan drug development and rare disease is a disease prevalence of less than 200,000 or less than 7.5 in 10,000 (Figure 3). For the EU EMA, the disease prevalence requirement is less than five in ten thousand. For both the US and the EU, if the disease prevalence is higher than the stated guidelines, it will need to be demonstrated that there are insufficient incentives to generate return (i.e. without incentives, the drug would not generate sufficient return).
In addition, while not a requirement in the US, the EU requires that it be shown how the drug will treat a life-threatening or seriously debilitating or chronic condition, which is a particular point that will need to be addressed if looking at development in both regions.

In the US, if the disease prevalence is higher, Vonderhaar explained that there needs to be “enough information to establish a medically possible basis that the drug will be effective” – this may be achieved by using preclinical data, although clinical data always lends the greatest strength to a case. In addition, if there is a drug that already has marketing approval for the same indication, you will need to demonstrate superiority of your drug over the available therapy in a superiority trial (or demonstrate intent to conduct one) – this must be included in the orphan drug application.
It is important to note that orphan drug applications do not go through the IND and instead go through the office of orphan drug products at the FDA, which involves a different submission process.
From the European perspective, the EMA requires a satisfactory method of diagnosis, prevention or treatment and non-similarity to other orphan products for the same indication.
Overall, understanding how to approach development from a global perspective, if you’re looking at more than one region, is something that should be considered early in development.
Features of Orphan Designation in the US vs. the EU
For orphan drug designation, the US FDA gives market exclusivity of seven years and a New Drug Application (NDA)/Biologics License Application (BLA) is not subject to user fee and a tax credit. With the EMA, the market exclusivity is ten years and also features fee reductions, direct access to centralized procedure, local national incentives and separate licenses for orphan and non-orphan. Despite some of these differences, understanding the key features of orphan designation in both the US and EU is key to moving programs forward.
Timelines are always critical in the development process so planning ahead is essential. The EMA website outlines these timelines and being aware of, and up to date on them, is important, because they do tend to change .
When it comes to orphan drug designation, a pre-submission meeting prior to filing the application is advisable. This is important because it can help address any issues that could occur, which can be outlined by officials at the EMA.
FDA Meetings
Seeking scientific advice from the US FDA is instrumental in understanding whether you meet the requirements for potential designations and/or pathways. This is so you can ensure that you have alignment with the FDA with respect to a plan moving forward.
There are four different types of formal meetings with the FDA which can be used to seek scientific advice or other information: Type A, Type B, Type B End of Phase and Type C.
Type A meetings are 30-day meetings meaning that the time from submission to the meeting itself is 30 days. Types of meetings that fall into this category include dispute resolution, discussing clinical holds, special protocol assessments and then meetings within 30 days of FDA issuance of a refuse to file (RTF) letter. Rare and orphan diseases typically do not fall into a type A category unless you come to face one of these situations.
The Type B 60-day meeting is one of the most common types of meetings. This entails that you have 60 days from submission of the meeting request letter to the meeting itself, with the briefing package due 30 days prior to the meeting. Most meetings related to pre-IND meetings, which are very common for rare and orphan diseases, fall into this category. This type of meeting will help ensure that there is alignment with the agency because you may be in more expedited development. Other types of meetings that fall into this category, and particularly important for rare and orphan diseases, are products that have been granted breakthrough therapy designation, post action meetings for complete response letters or meetings to discuss run strategies.
The Type B End of Phase meeting is 70 days and can include both End of Phase I and End of Phase II meetings. The briefing package is due significantly earlier than for other types of meetings to ensure that the agency has sufficient time to review for these critical milestone meetings.
Type C meetings are 75 days long from submission of the meeting request letter to the meeting itself and includes any other topic or any other meeting with the FDA. For example, if you’re outside of a major milestone but looking to address any particular CMC preclinical or any other clinical or clinical pharmacology issues that are not included in an End of Phase or a pre-IND meeting, it will fall into the type C category.
There are also different formats of meetings with the FDA, namely written response only, teleconference and face-to-face. While you may request a certain type of meeting, the FDA has the final decision-making power in regards to the format of the meeting.
Scientific Advice
Scientific advice is very important from both a regulatory perspective and an operational perspective. From a regulatory perspective, it helps to ensure there is alignment with the FDA on the development strategy leading to fewer FDA queries and roadblocks. Advice will also help ensure that the proposed clinical development plan supports the marketing application and therefore having alignment on this before the marketing stage is important.
Vonderhaar said that the ultimate goal in these expedited accelerated pathways and designations is to “achieve the designations and use them to help speed up development or reduce the required clinical data to support an accelerated approach.” From an operational perspective, having scientific advice meetings ensures that there’s a lower risk of clinical hold and ensuing delays in study startup with fewer protocol amendments if there is greater alignment upfront with the FDA on things like dose, inclusion/exclusion criteria, endpoint safety monitoring and other protocol considerations. Early awareness and greater understanding also help overcome any potential challenges with respect to availability of the investigational product in case there are any manufacturing issues to avoid potential delays in the development program.
Regulatory Pediatric Considerations
For pediatric studies in the US, a study plan is due 60 days after the end of a Phase II meeting and before initiation of Phase III trials (if an End of Phase II meeting is not held). It should include CMC, non-clinical and clinical considerations. Referrals or waivers can be requested and with a waiver, the application is shortened. A six-month patent extension period is available for rare or orphan indications with large numbers of pediatric patients. Enrolment of older age groups in adult trials is something that should be considered and discussed with the FDA as part of a scientific advice meeting either at the end of a Phase II meeting or at an appropriate time in development.
In the EU, for a pediatric investigation plan (PIP) that is shortened, understanding the waivers, deferrals and the age groups involved is important. Timelines can be eight to 12 months for a PIP to be agreed upon, so managing and working within these time frames is critical.
Requesting scientific advice in preparation of a PIP is highly advisable and is free of charge. For the meeting, Daniel explained that you need to have a set of specific questions that you would like to have answered. He also advises to refer to the Q&A section on the EMA website because it provides a number of details including guidance on how to organize a PIP.
Accelerated Therapies for Patients
Accelerated designation programs and pathways for orphan drugs for rare and pediatric conditions are designed to assist in the rapid approval of treatments for diseases that may have limited, or no therapies at all currently. Medpace’s informative webinar highlighted key regulatory strategies and considerations in rare disease clinical drug development in both the US and the EU.
While there are some notable differences in the programs and program details between the two regions, there are also similarities, the most important of which include early engagement with regulatory agencies to gain guidance and scientific advice for development plan applications, and being aware of associated timelines. These strategies and considerations will help ensure a robust and seamless development process, which is key to delivering effective treatments to patients as quickly as possible.
This article was created in collaboration with the sponsoring company and the Xtalks editorial team.




Join or login to leave a comment
JOIN LOGIN