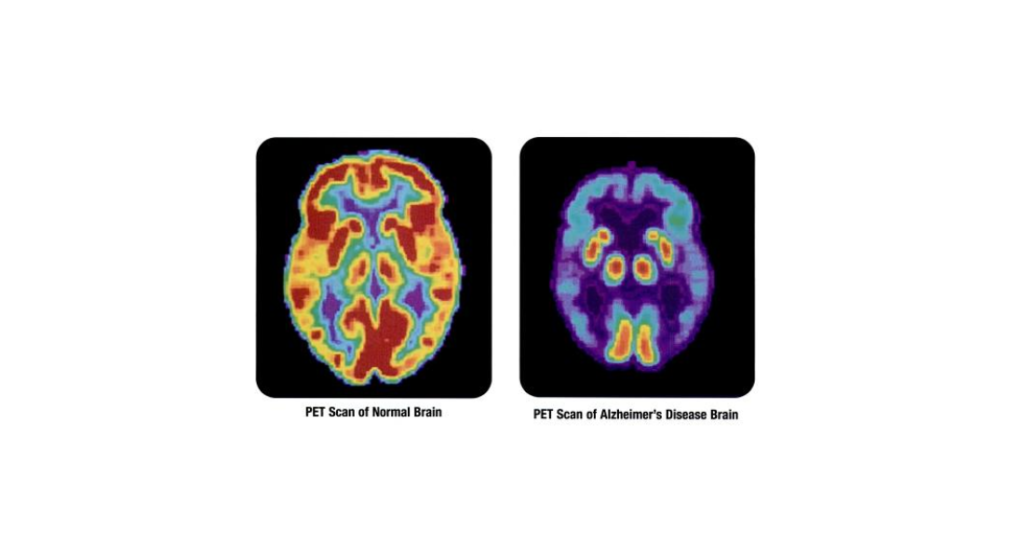
Brain biomarkers including amyloid-beta plaques and tau protein tangles are hallmarks of Alzheimer’s disease, however not all patients displaying this pathology go on to develop dementia. Now, researchers from The University of Texas think they’ve uncovered a potential mechanism explaining this phenomenon, and have published their theory in the Journal of Alzheimer’s Disease.
Though the cause of Alzheimer’s disease is not fully understood, it’s thought that the accumulation of amyloid-beta and tau proteins in the brain play a role in neurodegeneration characteristic of this type of dementia. While over five million people in the US are affected by Alzheimer’s, many more display this brain biomarkers of the disease without ever suffering cognitive decline.
“In previous studies, we found that while the non-demented people with Alzheimer’s neuropathology had amyloid plaques and neurofibrillary tangles just like the demented people did, the toxic amyloid-beta and tau proteins did not accumulate at synapses, the point of communication between nerve cells,” said Giulio Taglialatela, director of the Mitchell Center for Neurodegenerative Diseases. “When nerve cells can’t communicate because of the buildup of these toxic proteins that disrupt synapse, thought and memory become impaired. The next key question was then what makes the synapse of these resilient individuals capable of rejecting the dysfunctional binding of amyloid-beta and tau?”
RELATED: Looking Past the Amyloid-Beta Hypothesis Could Ring in a New Era of Alzheimer’s Research
Using high-throughput electrophoresis and mass spectrometry, Taglialatela and his colleagues studied frozen brain tissue samples from individuals who were participants in brain aging studies while they were still alive. Specifically, they looked at the protein composition of synapses to determine whether differences exist between individuals with Alzheimer’s disease, those with brain biomarkers of dementia without the symptomology, and people who exhibited no signs and symptoms of Alzheimer’s.
The researchers found that brain samples from those individuals with the dementia biomarkers but without Alzheimer’s were, indeed, unique when compared to the other two study groups. Their synaptic protein signature set them apart from patients with and without Alzheimer’s, which could explain why these individuals did not develop dementia despite having a build-up of amyloid-beta and tau in their brains.
“We don’t yet fully understand the exact mechanism(s) responsible for this protection,” said Taglialatela. “Understanding such protective biological processes could reveal new targets for developing effective Alzheimer’s treatments.”
While the current findings could prove helpful to drug developers looking for new ways to slow – or even stop – progression of Alzheimer’s disease, the amyloid-beta hypothesis itself is under question. Earlier this year, researchers from the NYU School of Medicine suggested a new mechanism for how Alzheimer’s disease is developed which involves a faulty cellular waste disposal system in the brain. Regardless of the exact cause of Alzheimer’s disease, drug development in this area is among the most challenging, with decades of failed late-stage trials prompting major drugmakers like Pfizer to exit the race to develop an effective treatment altogether.






Join or login to leave a comment
JOIN LOGIN