
Introduction
A First-in-Human (FIH) trial is the first step in testing novel treatments in humans. This is the first opportunity for the Sponsor to observe or “translate” all the observations from a myriad of preclinical tests in animals (or in test tubes) to people. A FIH trial tests the safety, side effects, optimal dosing, and dosing interval of a new treatment. It may also test the best route of administration for a new treatment (for example, by mouth, intravenous infusion, or injection) and how the treatment effects the body. The dose is usually increased in an orderly fashion in order to find the highest dose (maximum tolerated dose or MTD) that does not cause harmful side effects.[1] Typically, the subjects that are enrolled into these trials are known as “Normal Healthy Volunteers.”[2] However, due to different regulatory pathways and ethical concerns, FIH trials for rare disease indications and for anti-cancer therapeutics are the exception to this rule.
During this stage of testing, the cause of a clinical trial failure could be as simple as human subjects not responding to the drug the same way that animal models suggest.
Phase 1 clinical trial failures can also result from an insufficient understanding of how the investigational product interacts with the body (i.e., pharmacokinetics and pharmacodynamics). These interactions can vary (sometimes widely) between animals and humans, between healthy subjects and patients, and between one demographic group and another. For these reasons, how the Phase 1 trial is designed, carried out, documented, and reported are vitally important steps in the eventual commercial success and patient outcomes for years to come in that drug’s lifecycle. [3][4][5]
Designing the FIH Trial
Deep First-in-Human experience can yield effective study design and a protocol which includes all the parameters that will focus on efficiency, data quality, and shorter timeframes. Once the protocol is accepted by the FDA through the form of an Investigational New Drug (IND), an experienced Phase 1 Team can streamline the conduct of the study.
If you are opting to outsource the conduct of your FIH trial to a Contract Research Organization (CRO) or to a standalone clinical pharmacology unit (CPU), one of the cardinal considerations in the selection process is assessing the CRO/CPU’s experience in Phase 1 trial execution.
When selecting a partner to oversee your FIH trial, Sponsors should ensure that they are incorporating appropriate adaptive modifications into the trial design such as:
- Sentinel dosing
- Sample size adjustment
- Changing of the randomization fraction:
- to favor certain treatments
- to avoid unfavorable treatments
- Addition of special patient populations
- Addition of specific ethnic populations
- Addition of new treatment arms
- elimination of other treatment arms
The ability to effectively communicate the benefit of the above strategies is an important part of the process, ensuring that Sponsors can fully leverage proposed options in their program.
The diversity of new therapeutic entities (biologics & small molecules) that require FIH testing has required increased levels of vigilance over the past several years. Single and Multiple Ascending Dose designs often include multiple objectives:
- Safety and Tolerance
- Pharmacokinetics (PK)
- Pharmacodynamics (PD) [early indicators of “on-target” or “off-target” activity relative to drug exposure utilizing specific “Biomarkers”]
- Food effect
- Assessment of the effects on subsets of subjects based on age, gender or ethnicity
- Potential interactions with expected co-therapies
- Therapeutic outcome in a small panel of patients suffering with the targeted illness
Single Ascending Dose
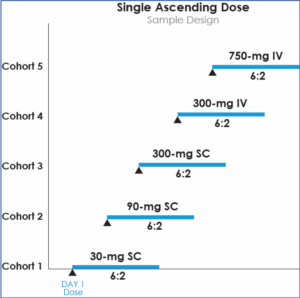
In Single Ascending Dose (SAD) studies, small groups of subjects are given a single dose of the drug while they are observed and tested for a period of time to confirm safety.[6][7] Typically, a small number of subjects, usually two, are entered at a particular dose as sentinel subjects.[2] If they do not exhibit clinically-relevant adverse side effects, the rest of the group is dosed. If unacceptable or unanticipated toxicity is observed in any of the subjects, additional subjects, usually two, are treated at the same dose.[2] This is continued until pre-calculated pharmacokinetic safety levels are reached, or intolerable side effects begin manifesting, at which point the drug is said to have reached the Maximum Tolerated Dose (MTD). If an additional unacceptable toxicity is observed, then the dose escalation is terminated and that dose, or perhaps the previous dose, is declared to be the MTD. This design assumes that the MTD occurs when approximately one-third of the subjects experience unacceptable toxicity. Variations of this design exist, but most are quite similar.[2]
Multiple Ascending Dose
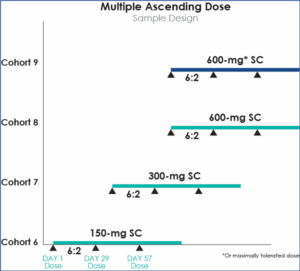
Multiple Ascending Dose (MAD) studies investigate the pharmacokinetics (described as what a drug does to the body), including receptor binding, post-receptor effects, and chemical interactions. Drug pharmacokinetics determines the onset, duration, and intensity of a drug’s effect and pharmacodynamics (defined as the response of the body to the drug). It refers to the relationship between drug concentration at the site of action and any resulting effects, specifically, the intensity and time course of the effect and adverse events, looking at safety and tolerability. In these studies, a group of subjects receive multiple low doses of the drug while samples of blood, and other bodily fluids, are collected at various time points and analyzed to acquire information on how the drug is processed within the body. The dose is subsequently escalated for further groups, up to a predetermined level.[6][7]
Study Design Impacts Resourcing Needs and Cost
There are several factors which will influence the resourcing needs and expected cost to run your FIH project. One of them is the size of the cohorts being contemplated for the study. In the conduct of Phase 1 studies, there are economies of scale in being able to run larger cohorts. As the cohort size gets larger, the per-subject staffing fees should decrease, as the costs are spread across a larger number of subjects. If sentinel dosing is incorporated into your protocol, you will need to factor in the additional 24-hour period (and personnel) that will be necessary to observe dose responses in the first two subjects at each dose level enrolled before proceeding to dose the remainder of the cohort. Multiply this factor by however many cohorts will incorporate sentinel dosing.
For example, view the below diagram for a sample schedule of Day 1 activities for a SAD study which incorporates sentinel dosing in the first six cohorts of dose escalation. The procedures and timepoints outlined in red will occur for the first two subjects, then repeat on a separate day for the remainder of the subjects in the cohort, effectively doubling the amount of dosing and PK collection days in the SAD cohorts.

Another strategy which will influence overall staffing needs, though not always considered, is the use of “Alternate” subjects in each cohort. Because enrolling full cohorts is of the utmost importance in FIH trials, and under-enrolling by even one subject may cause significant delays, experienced Phase 1 teams recommend checking in additional subjects on Day -1 (the day prior to dosing) to ensure that the full cohort can be dosed (e.g. Checking in 10 subjects to ensure a full cohort of 8). These additional subjects will mitigate the risks associated with subjects who drop out unexpectedly (due to personal issues, lab results, etc.). While you can expect additional resourcing needs and increased costs associated with identifying these subjects and performing all necessary procedures prior to dosing, these costs are far less than those caused by delays incurred by not dosing a full cohort.
Finally, there will be variability in cost and staffing needs when incorporating special populations such as specific ethnic groups, age groups, or patient arms into your FIH trial. It is reasonable to expect that subject recruitment timelines will need to be increased, and that more effort will be required when enrolling a population that differs from the standard “Normal Healthy Volunteer” (NHV). Incorporating a special population may also require the use of smaller cohorts to accommodate for the slower subject accrual rate. Consider some of the below factors when developing a recruitment plan for a FIH trial incorporating special populations:
- Gender stratification: Typically, it is easier to recruit male volunteers and there may be additional evaluations necessary for female volunteers (e.g. pregnancy tests, pap smears, etc.). Expect additional effort required if women of child-bearing potential (WOCBP) will be excluded.
- Ethnic populations: There may be language barriers and other cultural sensitivities which will need to be considered when creating subject-facing materials. Will “matching” criteria be required? If so, the cadence of recruitment and enrollment tactics could be impacted.
- Patient arms: May require additional consultative time from practicing physicians to confirm subject eligibility. Furthermore, if study eligibility criteria differ significantly from the standard of care, you will likely need to factor in additional time to identify subjects which meet all eligibility requirements.
Whether the trial is being managed internally or through outsourced partners, pay keen attention to cost and staffing efficiencies as sizes scale up. When the budget is produced, the number and assumed size of cohorts should be clear, as well as the resulting staffing needs associated with each. It will be important to identify those areas which are directly tied to the number of subjects in a cohort versus items which are fixed costs. This will provide true visibility into how the budget might be impacted if the number of cohorts or the subjects in each cohort fluctuates.
Safety and Risk Assessment
Safety is paramount in Phase 1 studies as these trials are usually only based on pilot studies in animals regarding potential pharmacologic and toxicologic barriers. However, because these barriers have yet to be studied in humans, there is significant benefit in using an experienced, 24-hour surveillance unit to host your FIH study.
Regulatory Agencies worldwide have produced guidelines for the biopharmaceutical industry in order to ensure that the safety and wellbeing of trial participants is always the priority. These guidelines place emphasis on the Sponsor’s responsibility to define the uncertainty associated with the treatment tested at each phase of its development and to describe how the potential risks that might arise from this uncertainty will be addressed within the design and conduct of the trial. The approach must be supported by well-documented scientific rationale from the outset and be responsive to data emerging over the course of the trial itself.
Working with Phase 1 teams who have formal written processes helps mitigate risk. This includes the providing counsel on the potential risks inherent in the study. Not just envisioned risks, but risks observed from the lessons learned in multiple previous trials of similar nature. This should include contingency plans, open and frequent communication processes, and robust quality and regulatory controls to ensure compliance and subject safety.
Prior to beginning a FIH study, it will be imperative to design and implement a thorough Safety Monitoring plan. This plan will outline the methods for data collection, frequency of data review, and clear recommendations for how to proceed with the overall study. An important component of the Safety Monitoring Plan will be the manner which Safety Review Meetings between cohorts are conducted.
Safety Review Meetings may be conducted in various ways depending on the expected risk of the investigational product. If low risk, decisions to proceed with dose escalation may center around the occurrence of Adverse Events (AEs) and can usually be performed by the Sponsor and the trial’s Principal Investigator(s), along with supporting members of the study team such as the Medical Monitor and Project Manager who can utilize data generated by the Data ManagementTeam. If the anticipated risk is moderate to high, third-party experts in the relevant therapeutic area may also be enlisted. During safety review meetings, several items are reviewed and considered when making the decision to continue with additional cohorts (at higher dose levels) such as:
- Subject demographics
- Adverse Events, including severity and relatedness to study drug administration
- Use of Concomitant Medications
- Trends or abnormalities in vital signs
- Clinical Laboratory Evaluations
- Physical Examinations
- The occurrence of dose escalation or individual stopping criteria
Aligning processes to assist Sponsors in managing safety risks in FIH studies requires years of experience in conducting Phase 1 trials. In the earliest stages of developing the protocol, risk management must be a consideration. By making these plans upfront and assessing adherence as the trial starts, the chances of losing valuable “clinical momentum” during the study can be significantly reduced.
Providing robust quality and regulatory controls throughout the entire study will promote compliance. Quality assurance processes, including internal training programs, vendor auditing, validating software and establishing SOPs, indicate that the team managing the study is committed to delivering high quality. Again, if you are outsourcing this project, evaluating a CRO/CPU’s established processes towards ensuring the safety of study subjects will be another significant consideration during the selection process.
Furthermore, since Phase 1 trials are not intended to evaluate efficacy, there are ethical as well as medical risks. For FIH studies, there is essentially no prior experience with the investigational agent that can be used to educate trial participants about its risks and benefits in a truly informed manner. Phase 1 trials may pay higher subject stipends than later phase studies and usually involve healthy human subjects, but the risk of unforeseen adverse events may exceed the comfort level of a novice Principal Investigator. Therefore, it is of the utmost importance to implement detailed risk mitigation measures with the medical and clinical operations teams during the planning stages of a FIH trial.
Responsibilities of the Principal Investigator (PI)
The initial responsibilities of the PI are to ensure that the protocol’s inclusion and exclusion criteria are met and to consider whether the benefits of participation in the trial outweigh the potential medical risks. In addition, the PI must monitor the subjects’ compliance with study requirements.
The PI and their team must also ensure adherence to the protocol, compliance with regulatory responsibilities, and maintenance of data quality. Finally, the PI is responsible for updating trial participants of any new risks or benefits that evolve as the trial progresses.
There are several pertinent questions the PI should ask before enrolling any subjects. As the main concern of any study is safety and not simply meeting target enrollment, the PI is responsible for transmitting the risks and benefits of the study treatment to subjects, even if data are unclear or unknown. The PI must also remain up to date on risk and benefit information that may be reported in a variety of sources. In addition to data in the Investigators Brochure (IB), the PI should remain current on studies in the peer-reviewed literature.
Recruiting Trial Volunteers
Across all phases of clinical research (Phase 1 through 4), challenges in patient recruitment are often accounted for by a lack of awareness of clinical trials and a “skepticism” of the tested compound’s efficacy compared to existing treatments. Recruitment challenges in Phase 1, however, have their own additional inherent challenges. Perhaps the most notable of these is the “guinea pig” fear or “test subject” paradigm. As the study drug’s safety and tolerability profiles have not yet been established prior to a FIH study, potential subjects often worry it may pose a danger to their “normal” wellbeing. A lack of clinical incentive is another common factor, as most Phase 1 trials are performed in subjects with no medical necessity for the tested compound.
Therefore, effectively deploying a successful volunteer recruitment and retention campaign will be necessary during the planning and execution stages. For a FIH trial, this will involve an increased need to provide subjects with background on the drug and how to reasonably interpret results from animal models (with no previous human data available), as well as thoroughly communicating the measures that will be put in place to ensure their safety throughout trial conduct, including the decision-making process for dose escalation.
Beyond this, subjects will need to be coached on what to expect and how they can successfully fulfill study requirements. Even the most well-trained Phase 1 teams, with a track record of executing protocols efficiently, will not be able to overcome deviations caused by subjects who are non-compliant with study commitments.
Resources such as study orientations and reminder cards with schedules can go a long way towards ensuring compliance. Successful compliance can be enhanced by additional retention activities such as providing transportation to and from study visits and frequent communication between trial staff and subjects. The underlying mindset should be to alleviate as much burden from subjects and remove as many barriers to participation as possible. By doing so, staff will be able to focus on what’s truly important for FIH trials: generating high-quality and accurate study data.
Conclusion
Successful completion of a FIH study is an important milestone in a new drug’s journey on its regulatory path to the Pharmacist’s shelf. If carried out correctly, a solid foundation of knowledge about safety and exposure at several dose levels given over several days becomes available to the development team in a relatively short time period (6 to 12 months). The results from the SAD and MAD trials ultimately inform every stakeholder at the Sponsor organization:
- Discovery Scientists: First evidence that observations in animal models are similar in man
- Preclinical Safety: Validation of safety at exposures tested in animals to support full development
- Formulation Scientist: Results provide insight into adequacy of dosage form development strategy
- Clinical Development Team: Obtain initial evidence of exposure, activity, and toxicity relationships that can inform the Phase2 planning process
- Clinical Safety Team: Obtain initial data for safety monitoring planning
- Regulatory Affairs: – Obtain needed information to update Investigator Brochure in support of Phase 2 development
- Marketing: Begin to collect data in support of the Target Product Profile
Furthermore, opportunities to accelerate or enhance a compound’s chances for successful development may be gleaned from data derived from the SAD and MAD program.
This article was created in collaboration with the sponsoring company and the Xtalks editorial team.
References
- “NCI Dictionary”. National Cancer Institute. 2011-02-02.
- DeMets, D., Friedman, L., and Furberg, C. (2010). Fundamentals of Clinical Trials (4th ed.). Springer. ISBN 978-1-4419-1585-6.
- Wong C.H., Siah K.W., and Lo A.W. (2018). Estimation of clinical trial success rates and related parameters. Biostatistics. https://doi.org/10.1093/biostatistics/kxx069
- Thomas D. W., Burns J., Audette J., Carrol A., Dow-Hygelund C., and Hay M. (2016). Clinical Development Success Rates 2006–2015. San Diego: Biomedtracker.
- Hay M., Thomas D. W., Craighead J. L., Economides C., and Rosenthal J. (2014). Clinical development success rates for investigational drugs. Nature Biotechnology 32, 40–51. https://doi.org/10.1038/nbt.2786
- “Phases of clinical trials”. Canadian Cancer Society. 2017. Retrieved 1 February 2017.
- Elizabeth Norfleet, Shayne Cox Gad, “Phase 1 Clinical Trials”, in Shayne Cox Gad, Clinical Trials Handbook, ISBN 978-0-470-46635-3, 2009, p. 247









Join or login to leave a comment
JOIN LOGIN