
Rare disease clinical development is undergoing rapid transformation, fueled by advances in therapeutic science, increasingly adaptive trial designs, and evolving global regulatory frameworks. For sponsors targeting small patient populations, these shifts are creating both new opportunities and operational complexities.
While rare diseases collectively affect hundreds of millions of people worldwide, individual conditions often involve small, geographically dispersed patient groups. This reality continues to complicate development, as natural history data may be limited, disease progression can vary widely, and recruitment often depends on a small number of specialized research sites. Designing studies that generate meaningful evidence while remaining feasible in such settings therefore requires close coordination across clinical, regulatory and operational teams.
“Rare disease clinical development is undergoing rapid transformation.”
— Jackie Widmer
In a recent webinar, Ann Woolfrey, MD, Vice President in the Medical Department at Medpace, was joined by her colleagues Tanya Konovalenko, MPharm, RAC, Director of Regulatory Affairs and Jackie Widmer, Director of Clinical Trial Management, to explore three key trends influencing the future of rare disease research.
Their discussion explored how platform trial innovation, advanced therapies, and expedited regulatory pathways are redefining how rare disease programs are designed and executed.
Read on for insights into designing effective trial strategies, maintaining regulatory alignment and advancing next-generation therapies in ultra-small populations.
Customizable Platform Trials and Accelerating Development Timelines
Traditional randomized trials were designed for conditions with large patient populations, established care pathways and familiar endpoints. Rare disease development rarely benefits from these advantages. Patient groups are often geographically dispersed, phenotypes can vary widely even within the same diagnosis and specialized expertise may be concentrated in only a few academic centers.
These constraints have driven growing interest in platform trial designs, which enable multiple therapies to be evaluated within a single coordinated framework rather than through separate studies.
As illustrated in Figure 1, platform trials use a layered structure that separates shared infrastructure from intervention-specific components:
- Platform (Master) Level: An overarching protocol defining common eligibility criteria, governance, statistical methods and core operational systems.
- Intervention-Specific Appendix (ISA) Level: Individual therapy modules detailing the investigational product, treatment schema and intervention-specific endpoints.
- Integrated Components: Shared elements such as centralized data collection, safety monitoring, operational workflows and statistical oversight that support all interventions.
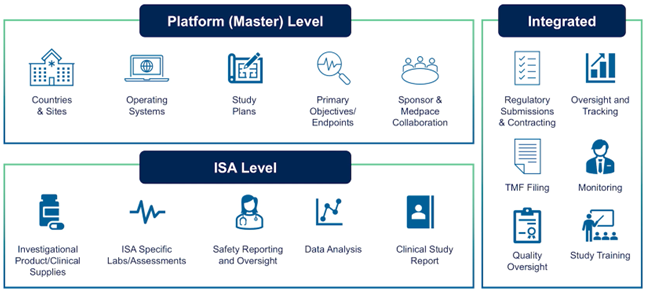
Figure 1. Platform trial structure enabling evaluation of multiple therapies within a shared master protocol.
This structure allows platform trials to remain active over extended periods as therapies enter or exit the program. Rather than launching a new study for each investigational product, sponsors can add therapies through ISA amendments, maintaining continuity within a unified research environment.
Key features of platform trials include:
- A shared master protocol governing all interventions
- The flexibility to introduce or remove therapies without restarting the study
- Standardized eligibility criteria and assessments that support consistent data collection
- Centralized infrastructure that reduces duplication
- More efficient use of limited patient populations, particularly important in ultra-rare diseases
- Opportunities to share control data across interventions in certain designs
“A platform trial can be designed to study multiple targeted therapies in the context of rare disease in a perpetual manner, with therapies allowed to enter or leave the platform,” Widmer explained.
Beyond operational efficiencies, platform trials can deepen scientific understanding of rare diseases. Standardized protocols and consistent data collection often generate valuable longitudinal datasets, which can help investigators interpret treatment responses and refine clinical endpoints in conditions with limited natural history information.
As highlighted during the webinar, this model can also reduce fragmentation across the research landscape. Instead of multiple sponsors or studies competing for the same small patient population, a shared platform enables more coordinated and efficient evaluation of therapies.
Platform trials require rigorous statistical planning, clearly defined governance and close coordination among sponsors, investigators and regulators. Master protocols must distinguish platform-wide elements from intervention-specific components, data analysis may occur at both the platform and ISA levels.
For sponsors advancing rare disease therapies, such a collaborative infrastructure can be highly valuable. Widmer noted that Medpace works closely with sponsors to design and operationalize complex studies, aligning scientific objectives with the realities of conducting trials in small, geographically dispersed populations.
In rare disease development, where patient numbers are limited and timelines are often compressed, platform trials offer a more durable clinical research environment than repeatedly building new trials from the ground up.
New Global Regulatory Frameworks for Rare Diseases
If platform trials represent one dimension of change in rare disease development, evolving regulatory thinking represents another. The traditional model of multiple large, randomized trials is often impractical, raising important questions about how to generate and evaluate substantial evidence for rare and ultra rare disease – where the global patient population may only be hundreds or sometimes just dozens of patients worldwide.
The FDA’s Rare Disease Evidence Principles (RDEP)
The FDA’s Rare Disease Evidence Principles (RDEP) framework was introduced to help address these challenges. Developed in response to expanding scientific and clinical activity in ultra-rare diseases, it recognizes that conventional development pathways may not be feasible for extremely small populations. Instead, the framework encourages sponsors and regulators to consider a broader range of evidence sources when evaluating therapies for rare genetic conditions.
“In September 2025, the FDA introduced the rare disease evidence principles, RDEP,” said Konovalenko. “This is not just a new guidance, it’s a commitment to a common sense approach for ultra-rare diseases with genetic defects.”
RDEP reflects a shift toward a totality-of-evidence approach. Rather than relying solely on large, randomized datasets, development programs may center on a single well-controlled clinical study supported by additional confirmatory evidence.
As illustrated in Figure 2, the RDEP framework emphasizes how different forms of evidence can be assembled to create a cohesive regulatory package. While a well-controlled clinical trial remains central, other data sources can strengthen the interpretation of treatment effects and help contextualize clinical outcomes.
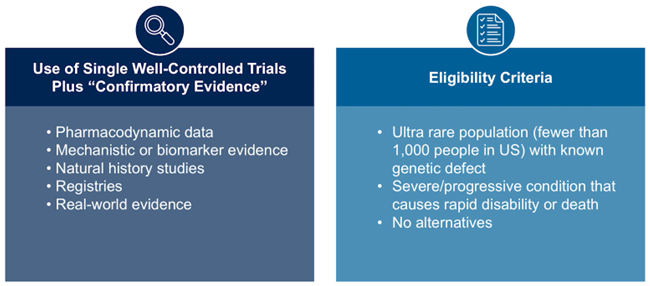
Figure 2. The FDA’s Rare Disease Evidence Principles (RDEP) framework allows one well-controlled trial supported by confirmatory evidence in ultra-rare diseases.
Together, these elements enable regulators to evaluate the robustness of a therapy’s evidence base despite limited patient numbers.
“To summarize, RDEP is not a lowering of standards; it’s a widening of lenses,” Konovalenko emphasized.
This distinction underscores the framework’s intent. The goal is not to reduce evidentiary rigor, but to demonstrate scientific validity through integrated data sources. In genetically driven diseases, for example, strong mechanistic rationale and biomarker responses may meaningfully reinforce clinical findings.
For sponsors, the implications are substantial. Development programs must be designed from the outset to integrate multiple evidence streams. Endpoint selection, availability of natural history data, quality of biomarker evidence and strategies for assembling confirmatory datasets all influence the credibility of the regulatory pathway.
In ultra-rare disease development, regulatory strategy and trial design are therefore inseparable. RDEP encourages a holistic approach to evidence generation, enabling therapies for small patient populations to be evaluated through frameworks that better reflect the realities of rare disease research.
Fast Track IND Processes and Incentives in China
Regulatory evolution in rare disease drug development extends beyond the US and Europe. China has introduced a series of policy reforms aimed at accelerating innovation in rare diseases and advanced therapies. These changes reflect a broader effort to shorten development timelines and strengthen incentives for treatments addressing significant unmet medical need.
A key development is China’s introduction of an expedited 30-day investigational new drug (IND) review pathway for certain innovative therapies. If regulators raise no objections within this window, sponsors may proceed directly to clinical trial initiation. This marks a meaningful shift from earlier review timelines, which were often considerably longer and could delay clinical development.
During the webinar, Konovalenko noted that these reforms are reshaping how sponsors position China within global development strategies. Faster regulatory timelines increasingly enable clinical programs in China to run in parallel with those in the US and Europe, rather than being introduced later. In addition to expedited IND review, China has introduced several policy mechanisms to address the “intellectual property gap” for orphan drugs that support rare disease innovation and improve the commercial viability of orphan drug development. The new policy that will take effect in May 2026 will formalize market exclusivity period for rare disease therapies preventing follow on competitors from entering the market. This will be in addition to existing a 6-year data protection period from the date of China marketing authorization which prevents competitors from using originator’s undisclosed clinical trial data to gain approval for generic or biosimilar versions of a new drug and financial incentives such as the 3% reduced VAT for imported orphan drugs.
Collectively, these updates signal an effort by Chinese regulators to create a more supportive environment for rare disease research and drug development. For sponsors, the implications are both strategic and operational. Accelerated IND timelines can reduce the time needed to activate sites and begin patient enrollment, but they also require development programs to be operationally prepared much earlier.
As a result, clinical operations, regulatory planning and supply chain logistics must be aligned well in advance of submission. Sponsors pursuing global rare disease programs may also need to coordinate development strategies across jurisdictions to ensure evidence packages remain coherent while meeting differing regulatory expectations.
For rare and ultra-rare diseases in particular, these reforms reinforce the importance of globally integrated development strategies. Patient populations are often widely dispersed, and recruitment may depend on activating sites across multiple regions. As China’s regulatory framework continues to evolve, the country is increasingly becoming a core component of international rare disease development programs rather than a secondary market addressed later in the product lifecycle.
Ex Vivo Gene Therapy for Rare Inherited Diseases
One of the most scientifically intensive areas of rare disease research is ex vivo gene therapy for inherited disorders, particularly those affecting the hematopoietic and immune systems. In these approaches, patient cells are collected, genetically modified outside the body and reinfused following conditioning therapy. Once engrafted, corrected cells can repopulate the hematopoietic system and enable sustained therapeutic gene expression.
Ex vivo strategies are especially suited to single-gene disorders, where restoring gene function in hematopoietic stem cells addresses the underlying disease biology rather than only managing symptoms.
Ex vivo gene therapy approaches are currently being explored across a growing range of inherited diseases, including:
- Severe combined immunodeficiency (SCID)
- Wiskott-Aldrich syndrome
- Beta-thalassemia
- Sickle cell disease
- Metachromatic leukodystrophy
- Cerebral adrenoleukodystrophy
- Fanconi anemia
- Other rare inherited metabolic and immunologic disorders
These diseases share a common feature: genetic defects that impair hematopoietic or immune cell function, making them appropriate targets for stem cell gene modification.
As shown in Figure 3, gene therapy strategies include gene augmentation, gene correction and gene disruption. Although mechanistically distinct, all aim to restore normal cellular function through targeted genetic modification.
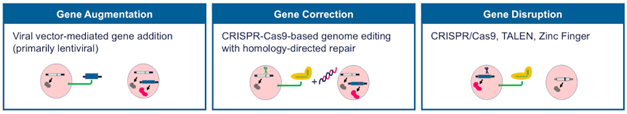
Figure 3. Major gene therapy strategies, including gene augmentation, gene correction and gene disruption.
A growing number of ex vivo gene therapies have reached clinical practice, demonstrating the translational potential of these approaches. Examples include:
- Zynteglo® for transfusion-dependent beta-thalassemia
- Skysona® for cerebral adrenoleukodystrophy
- Libmeldy® for metachromatic leukodystrophy
- Casgevy™, a CRISPR-based therapy approved for sickle cell disease and beta-thalassemia
Collectively, these advances show how modifying hematopoietic stem cells can deliver durable clinical benefit in diseases that previously required lifelong supportive care.
Ex Vivo Gene Therapy Clinical Study Design
Clinical development programs for ex vivo gene therapies differ fundamentally from conventional drug trials, as they integrate cell collection, advanced manufacturing and transplantation medicine within a single therapeutic pathway. Development timelines typically illustrate how each stage connects operationally and clinically, from patient screening and stem cell collection through conditioning therapy, engraftment and long-term follow-up (Figure 4).
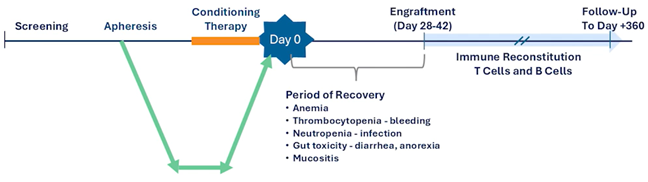
Figure 4. Clinical timeline for ex vivo gene therapy showing screening, apheresis, conditioning therapy, infusion (Day 0), engraftment, recovery period and long-term follow-up.
The typical ex vivo gene therapy workflow includes several sequential steps:
- Patient identification and eligibility screening
- Stem cell mobilization and collection, typically through leukapheresis
- Shipment of collected cells to a manufacturing facility
- Genetic modification of the cells using viral vectors or gene-editing technologies
- Product quality testing and release
- Cryopreservation and shipment back to the clinical site
- Patient conditioning, often using chemotherapy such as busulfan
- Infusion of the gene-modified cell product
- Post-transplant monitoring and long-term follow-up
Each step introduces specific clinical and operational considerations that can influence trial success:
- Eligibility screening must account for disease progression, genetic confirmation and clinical stability before manufacturing begins
- Adequate stem cell yield is critical for production
- Patients must remain clinically stable during manufacturing timelines
- Conditioning therapy adds further safety and monitoring requirements
- Extended follow-up is necessary to assess both safety and durability of response
Because each therapy is individually manufactured, close coordination among clinical sites, manufacturing facilities and logistics providers is essential throughout the study.
Challenges for Clinical Research Teams
While ex vivo gene therapies offer substantial therapeutic potential, they also introduce complex operational challenges for clinical research teams.
Recruitment and Screening Challenges
Rare disease trials already involve extremely small patient populations. In ex vivo gene therapy studies, recruitment may be further constrained by stringent eligibility criteria related to disease severity, organ function and transplant readiness, with key recruitment and screening challenges outlined in Table 1.
Table 1. Common recruitment and screening challenges in ex vivo gene therapy clinical trials.
| Challenge | Impact on Clinical Trials |
| Extremely small patient populations | Limits enrollment speed and geographic reach |
| Disease heterogeneity | Makes standardized eligibility criteria difficult |
| Rapid disease progression | Patients may become ineligible during screening |
| Limited specialized sites | Requires patients to travel long distances |
| Pediatric populations | Introduces additional ethical and logistical considerations |
These challenges can significantly affect timelines and require careful coordination between investigators, disease specialists and sponsors.
Hematopoietic Stem Cell Collection
Stem cell collection introduces further operational complexity. Leukapheresis requires specialized clinical expertise and may not be feasible for very young patients or those with certain complications. Alternative approaches, such as bone marrow harvest, may be needed, and backup collection procedures are sometimes planned to ensure sufficient cell yield for manufacturing.
Product Manufacturing and Gene Manipulation
Following collection, patient cells must be transported to a manufacturing facility for genetic modification, representing one of the most technically demanding stages of ex vivo gene therapy development.
As shown in Figure 5, tightly coordinated logistics networks connect clinical sites, couriers and manufacturing centers.
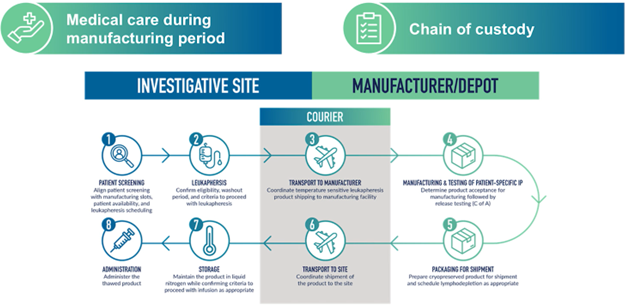
Figure 5. Chain-of-custody workflow linking investigative sites, couriers and manufacturing facilities during autologous gene therapy development.
Patient-specific products move through this system under strict tracking and temperature-control requirements, making maintenance of chain-of-identity and chain-of-custody critical. Successful execution depends on coordination among clinical investigators, manufacturing teams and logistics providers. Manufacturing timelines also introduce risk, as disease progression during processing may prevent patients from receiving the final product.
Transplant and Post-Treatment Considerations
The final therapeutic stage involves reinfusion of gene-modified cells following conditioning therapy, typically in transplant units experienced in hematopoietic stem cell transplantation.
Conditioning regimens, often busulfan-based, create bone marrow space for engraftment but also introduce toxicity risks requiring close pharmacokinetic monitoring.
Operational execution at this stage requires collaboration among transplant physicians managing conditioning and infusion, disease specialists assessing clinical outcomes, clinical trial teams overseeing protocol adherence and safety monitoring and medical monitors evaluating emerging safety signals across sites. Coordinating these perspectives is essential to protect patient safety while generating robust clinical trial data.
Future Prospects of Ex Vivo Gene Therapy for Rare Inherited Diseases
Looking ahead, ex vivo gene therapy technologies are expected to continue evolving rapidly in the coming years. Although several therapies have already received regulatory approval, the field remains highly innovative, with scientific progress improving both the biological effectiveness of gene modification and the operational feasibility of delivering these treatments.
Advances expected to shape the next generation of programs include improvements in cell-collection methods, more precise gene-editing platforms and new strategies to reduce treatment-related toxicity.
Key areas of progress include:
- Enhanced stem cell mobilization agents that increase hematopoietic stem cell yield during leukapheresis
- Next-generation gene-editing technologies, such as advanced CRISPR systems and alternative platforms designed to improve precision and reduce off-target effects
- Refinements in viral vector engineering to strengthen transduction efficiency and durability of gene expression
- Shorter, more scalable manufacturing workflows that may reduce the interval between cell collection and infusion
- Less-toxic conditioning approaches, including antibody-based regimens that selectively target hematopoietic stem cells while minimizing broader chemotherapy-related toxicity.
Together, these developments address major barriers in ex vivo gene therapy, particularly by improving manufacturing timelines and conditioning safety. Such progress may expand patient access while reducing operational demands on clinical trial sites.
Woolfrey emphasized that advancement in this field depends not only on laboratory innovation but also on operational expertise and the integration of clinical, regulatory and manufacturing strategies needed to transition therapies from experimental interventions to broadly available treatments.
As the field matures, gene therapy development programs are expected to become more efficient and scalable. Faster manufacturing timelines, safer conditioning regimens and more precise gene-editing tools could enable earlier intervention and reduce treatment-related risks. This is especially significant in rare inherited diseases, where early correction of genetic defects in hematopoietic stem cells may deliver durable disease modification rather than ongoing symptom management.
Continued progress will depend on close collaboration among scientific researchers, clinical investigators, manufacturing specialists and regulatory authorities. As Woolfrey noted, “At Medpace, we stay ahead of emerging trends by combining scientific insight, operational excellence and regulatory intelligence.”
Ex vivo gene therapy presents both promise and complexity for sponsors and clinical research teams. While scientific innovation is progressing rapidly, successful clinical translation depends on tightly coordinated execution across development stages. As the field evolves, these technologies are set to play a growing role in reshaping care for rare inherited diseases.
This article was created in collaboration with the sponsoring company and the Xtalks Editorial team.





Join or login to leave a comment
JOIN LOGIN