According to a recent review published in Nature, as of April 2022 almost 1,800 active cell therapy clinical trials were listed in ClinicalTrials.gov, a 33 percent increase from 2021.
The rapidly expanding landscape of cell therapy trials has put an operational strain on many clinical sites due to limited resources and infrastructure.
“In our experience, the successful operational execution of cell therapy trials involves complex regulatory, clinical and operational considerations, which are not required in trials using small molecules,” stated Dr. Jeff Vassallo, Senior Director of Clinical Trial Management, at the global clinical contract research organization (CRO) Medpace.
“These details must be thoughtfully considered early in clinical development to ensure studies are completed within the projected timeline and budget,” added Dr. Vassallo.
In a recent webinar, Dr. Vassallo discussed the operational considerations for complex cell therapy clinical trials. The webinar also included Dr. Ann Woolfrey, Vice-President, Medical Department, Medpace and Lauren Holton, MSN, APRN, FNP-BC, Advanced Clinical Practitioner, Medpace, who provided insights on how to overcome medical hurdles associated with cell therapy trials. In addition, Dr. Jan Ohotski, Principal Regulatory Submissions Technical Advisor, Medpace, offered insights on strategies to overcome regulatory hurdles in cell therapy trials.
Watch the free, on-demand webinar to gain insights from this group of experts on how to overcome common challenges associated with complex cell therapy trials.
A Survey on Global Cell Therapy Trials
Medpace recently evaluated the performance of their cell therapy trials in order to determine which factors contributed to study delays. Among 20 recent trials, 17 experienced delays in meeting key milestones. The most common factors associated with delays were insufficient non-clinical development and chemistry, issues with manufacturing and controls (CMC), clinical trial design problems, inadequate site and vendor feasibility, and regulatory authority submission delays.
Together, the data indicated a common pattern of delays with a root cause associated with siloed planning and decision-making originating with the Sponsor early in clinical development.
“Specifically, while the numerous stakeholders (such as Manufacturing, Operations, Regulatory Affairs, Clinical/Medical, and CRO/Vendors) and accompanying processes are typically considered in clinical development, the decisions involving the protocol and product design, in addition to operational nuances of clinical practice for the indication, are often considered independently, and not thoroughly integrated early in the process,” said Dr. Vassallo.
As a result of this siloed decision-making and planning, confirmation of suitability of the protocol and product design may occur much later during regulatory submission or patient enrollment, thereby requiring re-work, timeline delays and an increased budget.
However, this data also suggests that implementing a strategy of integrated, cross-functional planning, which leverages best practices early in clinical development, can enrich the likelihood of success when executing cell therapy clinical trials.
Paradigm Shift to Integrated Expertise
To help overcome the complex challenges of cell therapy trials, there needs to be a paradigm shift towards the integrated expertise of non-clinical development, regulatory/clinical trial design, and site and vendor feasibility early in clinical development.
Additionally, the protocol needs to be developed around the patient instead of the patient around the protocol.
Strategies to Overcome Regulatory Hurdles of Cell Therapy Trials
According to Dr. Ohotski, regulatory collaboration with Non-Clinical, Manufacturing, Clinical Operations and Logistics teams is crucial in designing the study, collating required data, establishing the manufacturing process, and setting up logistical arrangements including labeling, depots, and release processes.
It is important to have a comprehensive Product Development Plan (PDP) that is developed early and in collaboration with regulators and the non-clinical, manufacturing, clinical operations and logistics teams.
During the webinar, Dr. Ohotski reviewed the important considerations for the PDP, which include the following:
- Consider the regions and countries suitable for product development.
- This would then set the regulatory requirements for the Protocol Design, Non-Clinical/Clinical and CMC data.
- Engage with the Regulatory Authorities early on in these regions/countries.
- This helps shape the PDP and ensures Protocol Design, Non-Clinical/Clinical and CMC data are fit for purpose.
- Requirements for Phase I trials may not be as stringent as Phase III trials.
- It is good to consult with the Regulatory Authorities as the product moves through the development process, in line with Regulatory expectations.
- Discuss any changes to the manufacturing process and establish tests required for compatibility.
Global Regulatory Activities: Cross-Functional Collaboration Throughout Product Development
During the webinar, Dr. Ohotski presented the type of support that Medpace’s Regulatory Team can provide throughout product development, in the areas of CMC, Non-Clinical, Clinical/Medical and Labeling/Logistics (Figure 1).
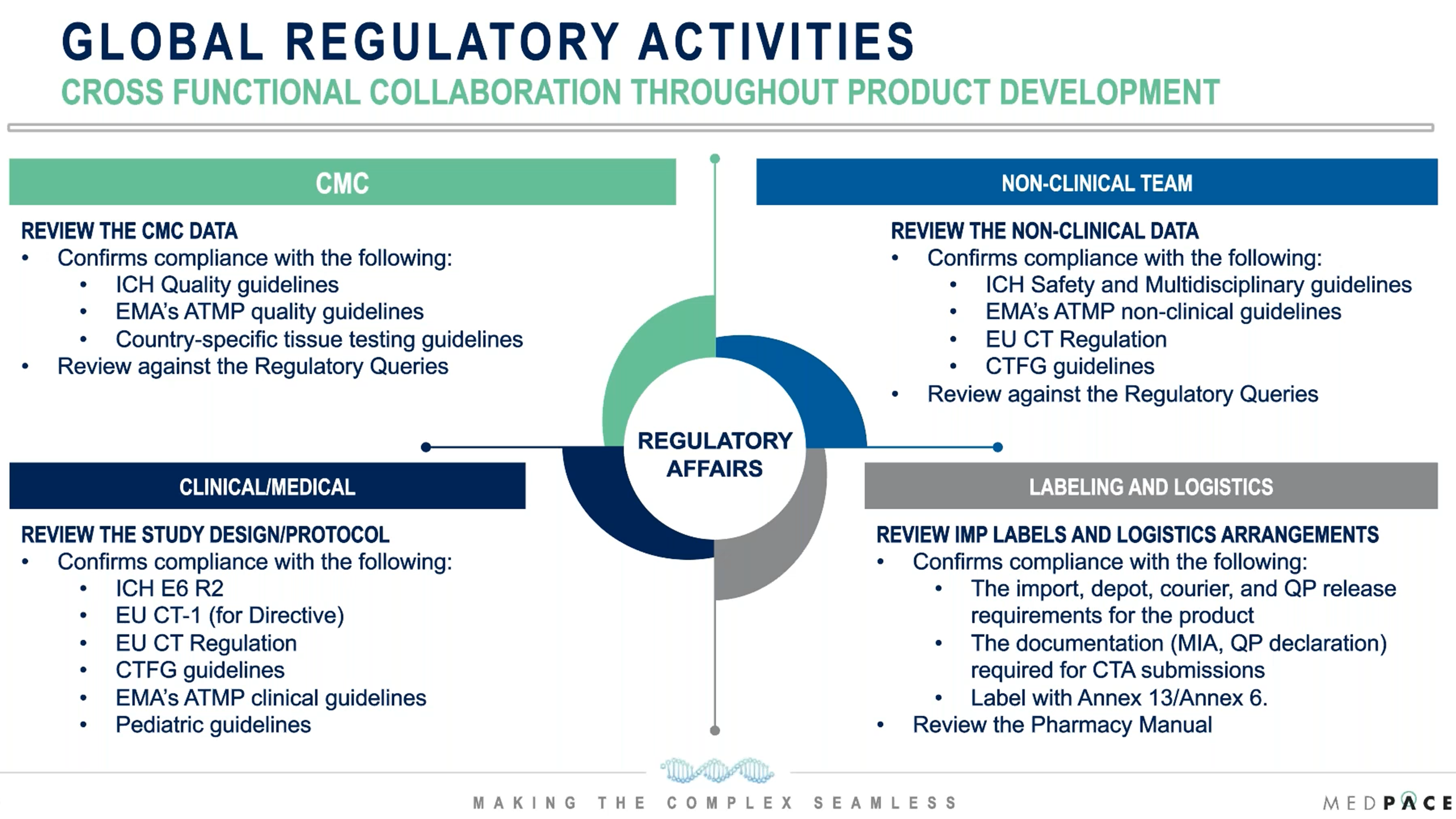
Figure 1. Medpace Regulatory Team support throughout product development.
“One of the main tasks of the Regulatory Team is to provide feedback based on the guidelines and regulations applicable to the product,” said Dr. Ohotski. “At Medpace, we go further by having a specialist regulatory technical team support our clinical trials, particularly for cell-therapy studies.”
Medpace Regulatory Expertise: Cell and Gene Therapy Regulatory Experts Embedded Within Operations
According to Dr. Ohotski, Medpace Technical Advisory Team supports Regulatory submissions for Advanced Therapy Medicinal Product (ATMP) trials in the following ways:
- Brings a wealth of hands-on experience with clinical trial operations for ATMP products.
- Extensive knowledge of regional and country specific requirements.
- Provides routine analysis of queries received in previous and ongoing trials via the Medpace Query Database. With this tool, the team can analyze trends and conduct lessons learned with a focus on the current regulatory landscape.
- Maintains document templates, checklists and delivers internal training as subject matter experts.
Medpace advanced therapy experts have extensive understanding of the Clinical Trial Authorization (CTA) requirements for ATMP trials in this rapidly evolving regulatory environment.
“Using the Medpace Query Database is a high value added to our operations because it enables us to stay on top of current requirements which may not be reflected in guidelines and regulations,” explained Dr. Ohotski.
Strategies to Integrate Clinical Considerations into Development of Cell Therapy Trials
The siloed nature of protocol development, set apart from CMC and regulatory considerations, often results in delays, clinical holds, and re-work, before the study can be operationalized. Dr. Woolfrey discussed key areas which an integrated approach could increase the potential for meeting milestones.
1) Non-Clinical Development Factors to Consider
The feasibility of translating a product from preclinical to clinical may not be fully known, particularly in first-in-human trials.
“Often our cell products initially derive from academic institutions and what was feasible in a single center study may not be commercially possible,” explained Dr. Woolfrey. “Therefore, CMC and the validation of methods of production and delivery should be done before commencement of the clinical trial so that these issues are all worked out.”
Dr. Woolfrey suggested that protocols incorporate feasibility criteria, which is particularly important for autologous cell therapy, but it is also important for off-the-shelf products, considering the feasibility of achieving target cell doses, the logistics of delivering the cells, and the capacity of sites to manage the cell product.
In addition, protocols should incorporate language that allows the trial to re-evaluate the manufacturing process at an early timepoint in the study, so as to not prolong a futile endeavor or discourage investigators and patients.
To help prevent delays in operationalizing the protocol during patient recruitment, Ms. Holton also recommended having practice sessions known as mock runs from vein-to-vein.
“During these mock runs, we will go through every step of the process at the site and with the staff members as if a patient were ready to be infused with a cell product. By doing this, we can identify potential pitfalls at the site level and mitigate these, so that when it comes time for a patient to be infused, the process is as seamless as possible,” explained Ms. Holton.
2) Clinical Trial Design Considerations
According to Dr. Woolfrey, institutional standards and workflow should be considered, as protocol activities might not coincide with institutional standards, especially if this is a global study.
The logistics of patient referral patterns and necessary patient support services should be considered.
“We frequently see that the schedule of events has a series of screening evaluations that may take place over a certain period of time, whether it’s two weeks or a month. But some of these evaluations can only be scheduled once the patient has signed the consent and is at the institution. Dr. Woolfrey noted that scheduling biopsies and imaging in academic centers typically requires pre-planning yet may not be allowed until the patient is registered at the site.
“For patients (especially patients with rare disease) who are not living in the area near the trial site but are referred from a different hospital, it may not be feasible for the patient to stay during that whole screening time, and it may discourage patients from joining the study” added Dr. Woolfrey.
Another factor for cell therapy clinical trials is to consider aligning treatment of underlying disease with protocol requirements (e.g., bridging therapies, prohibitive medications).
“It’s important to discuss protocol requirements with your key opinion leaders, the ones who are both knowledgeable about the disease indication, as well as the cell therapy processes at the sites that you’re considering,” added Dr. Woolfrey. There may be a large disconnect between the practices of the cellular therapy team and the patient’s primary physician (e.g., oncologist, rheumatologist) with respect to patient flow, scheduling of procedures, availability of clinical appointments, etc., which need to be understood before finalization of the protocol.
3) Site and Vendor Feasibility
As cell therapies are expanding to a number of therapeutic areas, it is vital to have a collaborative approach between all expert groups and vendors to ensure that lessons learned and best practices are considered.
“It’s also important to identify investigator roles. Perhaps the best approach is to have group experts be co-investigators or maybe sub-investigators,” said Ms. Holton.
4) Regulatory Authority Submission
An example of how the medical and regulatory teams can interact is during safety monitoring. According to Dr. Ohotski, safety parameters for first-in-human studies can be assumed to be relatively conservative.
“Regulators often have a great concern regarding safety monitoring between patients, looking at dosing schedules and observations performed prior to dosing the next patient, especially in early phase clinical trials,” said Dr. Ohotski. “It is critical to be conservative with dosing of the initial set of patients and with the duration of safety monitoring in between patients. It should allow enough time to fully assess the safety before the next patient is dosed.”
Strategies to Overcome Operational Hurdles in Cell Therapy Trials
During the webinar, Ms. Holton identified the following key strategies to help overcome the operational challenges of cell therapy clinical trials:
- Execute a manufacturing feasibility study.
- Integrate clinical workflow considerations in the packaging and handling design of the investigational product (IP).
- Develop the IP manufacturing and storage strategy to include logistical considerations.
- Ensure the screening schedule and eligibility criteria are realistic for the disease indication and necessary for the IP.
- Determine the support a site will require in terms of infrastructure and clinical support, especially if cell therapy is not common in the chosen indication or region.
“We need to consider how much support a site may require. Some sites may have very little experience in participating in cell therapy trials, so it’s key to assess this upfront and find out what experience level a site has during study start-up,” said Ms. Holton.
Medpace Cell Therapy Experience Applied
Recognizing the complexity of cell therapy studies, Medpace evaluates the following key variables during the request for proposal (RFP) and study setup phase, to determine study complexity and to identify risks requiring mitigative plans:
- The cell product manufacturing process.
- The number and experience of the vendors involved in the cell tracking system.
- The protocol study design and regulatory considerations.
- The global reach of the study.
- The indication and corresponding cell therapy experience of the appropriate clinical sites
- Oncology clinical sites are typically FACT (Foundation for the Accreditation of Cellular Therapy) or JACIE (Joint Accreditation Committee ISCT-Europe & EBMT) accredited
- Sites in other therapeutic areas may not have accreditation, and consideration must be given to whether it is required in order for them to participate.
- Do sites in non-oncology therapeutic areas need additional training or infrastructure to support the studies?
- The handling and infusion requirements of the cell product and expected adverse events.
“Medpace critically assesses all cell and gene therapy RFPs, utilizing an integrated, cross-functional expertise to ensure appropriate regulatory, clinical and operational considerations are included as part of the clinical development strategy,” said Dr. Vassallo.
Medpace’s Experience in Conducting Cell Therapy Clinical Trials
To date, Medpace has conducted more than 130 cell and gene therapy studies, including Phase I, II and III studies, in 30 countries and across different therapeutic areas (Figure 2).
Medpace has IP experience with both autologous and allogeneic cellular products, ex vivo and in vivo gene transfer, and a number of different gene-editing mechanisms.
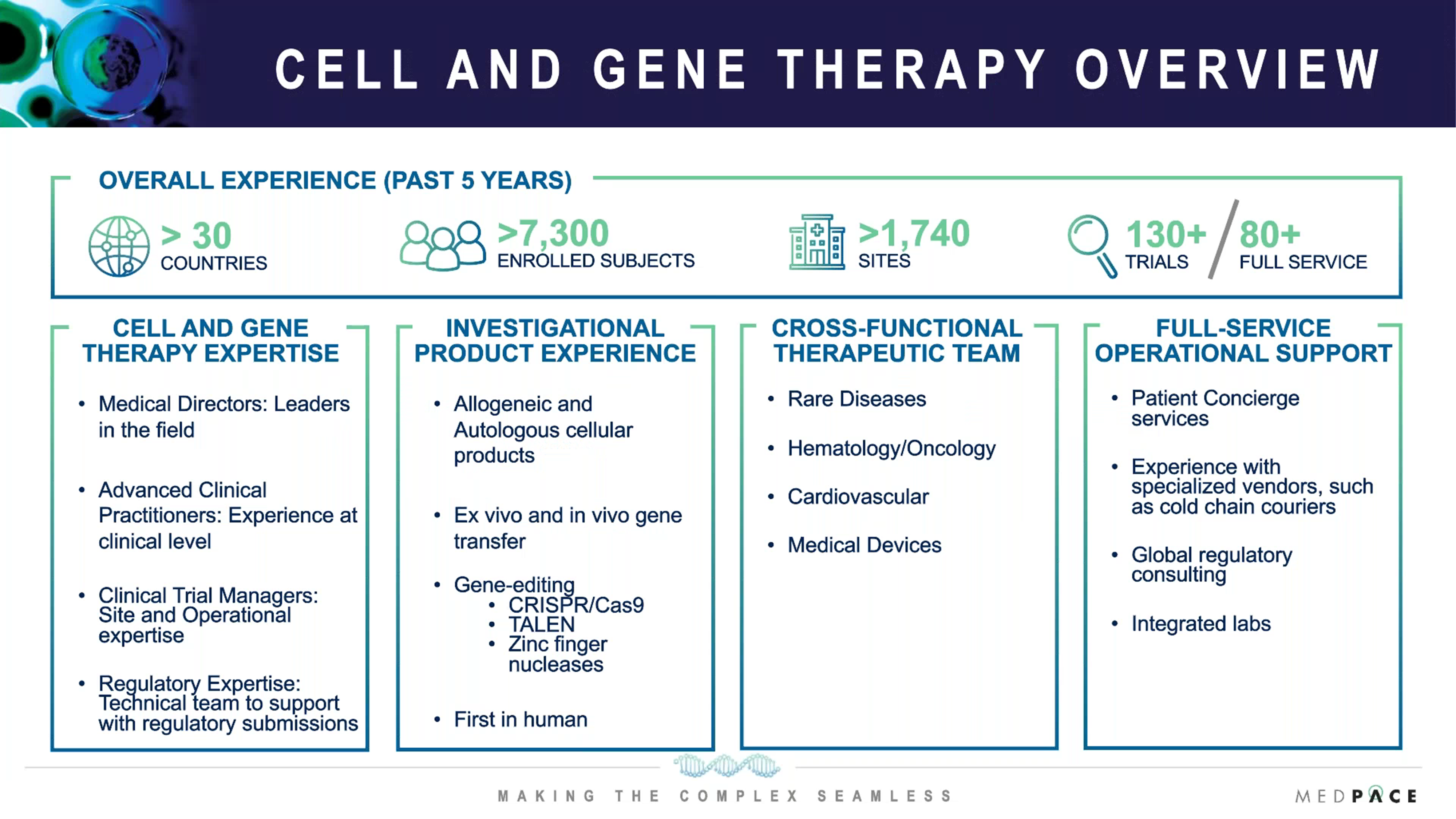
Figure 2. An overview of Medpace’s experience in conducting cell and gene therapy trials.
“Medpace leverages our experienced nurse practitioners’ boots on the ground clinical and logistical knowledge to streamline these complex cell therapy studies,” said Ms. Holton. “For instance, I’ve utilized my expertise to assist study teams with all things IV infusion-related, ranging from questions about sites for administration, factors that may impact infusion times, drip rate, all the way to supplies that are consistent with those needed from compatibility testing.”
Dr. Vassallo emphasized that Medpace’s success in operationalizing these trials is predicated on a full-service model, which includes a cross-functional team of experts.
Frequently Asked Questions About Complex Cell Therapy Trials
Medpace’s recent webinar had a Q&A session where Dr. Woolfrey, Dr. Vassallo and Dr. Ohotski answered the following questions about common challenges associated with complex cell therapy trials.
Recognizing the need for integrated expertise early in clinical development, at what stage should a small biotech initiate collaboration with Medpace?
Dr. Ohotski: As early as possible, ideally when you are working on non-clinical studies and CMC. We can bring a wealth of knowledge in these areas, coordinating interactions with regulators and enhancing the product development plan.
Should cell and gene therapy studies only be conducted at FACT and JACIE-accredited sites? If so, why?
Dr. Woolfrey: The first cell therapies, the CAR T-cell therapies, were developed out of stem cell transplant centers to treat the diseases that those investigators were already taking care of, so there was no conflict there. All of the stem cell transplant centers are typically JACIE or FACT-accredited.
However, cell therapies are now becoming more focused in targeting diseases such as autoimmune diseases or rare diseases that are not typically overseen by investigators in the hematology/oncology field. These investigators may not have the experience either in delivering cells or monitoring for safety and their sites may not be JACIE or FACT-accredited.
Conversely, cell therapy experts in the FACT and JACIE-accredited sites may not understand the nuances of the rare or autoimmune disease under investigation so this ultimately must be a group effort involving sites with both areas of expertise.
The discussions between both disease experts and cell therapy experts does need to start early and, in our experience, it varies between institutions which group takes primary responsibility for the patient. Often it mimics what we do in our stem cell transplant programs where patients are referred to the program, monitored for safety for the first month or so, and then referred back to their disease physician. This process does require a dialog between disease experts and cell therapy experts and Medpace can facilitate those complex dialogs.
Are the operational delays and learnings applied to specific phases or therapeutic areas?
Dr. Vassallo: As part of Medpace’s experience to date across over 130 cell and gene therapy trials, I can say with confidence that we have identified trends which are not phase or therapeutic area dependent, instead we see these common pitfalls across all phases and therapeutic areas.
Part of the challenge going from non-clinical to Phase I and then to Phase II and III, and even on to commercialization, is that, as you increase the number of patients, the number of sites and the number of countries, you have increased variables, which may make it more difficult to operationalize these studies.
The rapidly growing landscape of cell therapy studies has put a huge burden on clinical sites, with an emphasis on FACT and JACIE-accredited sites. Therefore, consideration must be given to the cell therapy labs at those sites in order to determine if they have the adequate resources to support those studies.
How can sponsors be assured that an autologous cell therapy product can be manufactured for patients in a specific indication, when a protocol is developed based on non-clinical or healthy volunteer data?
Dr. Woolfrey: The way to assure this to have experience manufacturing the product using blood samples from the target patient population, however this is not always feasible, especially during preclinical development.
As we have observed, even developing these products from healthy volunteers may not give a strong idea about how feasible it will be in the disease population. However, the protocol development plan can be evaluated, and there should be a strategy for what to do if difficulties are encountered. Consideration should be given to how to handle the patients who might not achieve the target cell dose or if the target cell dose cannot be achieved in any patient. The data can be re-evaluated and reincorporated so that the study does not stop but moves forward with the new information.
Are there any other considerations at the site level which may contribute to delays in operationalizing cell and gene therapy trials?
Dr. Vassallo: This requires a risk management approach, and for each study we want to develop a comprehensive cell therapy manual which clearly outlines all stakeholders and all steps required to conduct the study. Once the cell therapy manual is developed, it is important to ensure all stakeholders understand their role in the study and the process they are responsible for operationalizing.
A mock run should be conducted at each site where the entire process is gone through. This would include all activities normally completed with the patient, the vendors and manufacturing sites, to ensure that the study product is delivered as intended with the appropriate quality, stability and in the timeframe expected. This way, everyone’s role as a stakeholder on the study is clearly understood and they can operationalize it seamlessly, without issues.
What we typically find is, that in every mock run that we conduct, there are key learnings in process and communication across stakeholders that we are able to modify, to ensure going forward, we have seamless process and communication.
The mock run becomes even more important when studies are conducted in multiple countries, and/or have manufacturing in one country and the autologous cell product is being shipped to another country.
To learn more about successfully managing complex cell therapy trials, register to watch the free on-demand webinar by Medpace.
This article was created in collaboration with the sponsoring company and the Xtalks editorial team.



Join or login to leave a comment
JOIN LOGIN