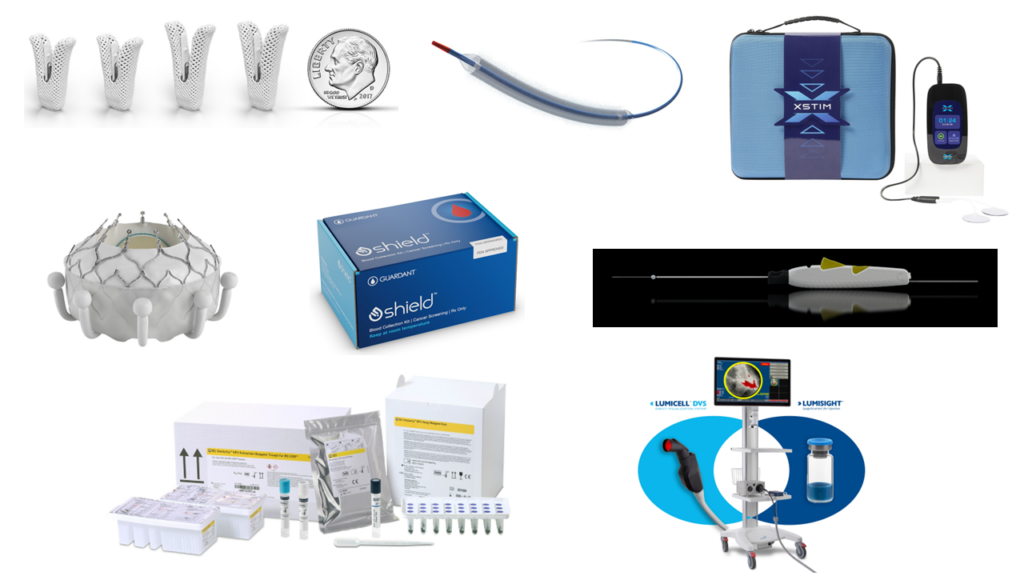
From non-invasive cancer diagnostics to life-saving cardiovascular implants, the latest medical devices cleared or approved by the FDA in 2024 reflect the remarkable strides in science and engineering. In this blog, we discuss some of these new medical devices of 2024 that have improved patient outcomes and enhanced quality of life worldwide.
1. TriClip G4 System
Manufacturer/developer: Abbott Medical
Date of FDA approval: April 1, 2024
Approved for: Tricuspid regurgitation (TR).

The TriClip G4 system, a transcatheter edge-to-edge heart valve repair system, is the first to treat TR. It is built on a tried-and-true clip-based technology. With four expandable size ranges, it offers a high level of adaptability, ensuring clinicians can select an implant size that best fits each patient’s tricuspid valve anatomy precisely and confidently. It comprises a clip implant, a delivery catheter and additional accessories. The clip is designed with arms that can open and close, enabling it to securely attach to the heart’s leaflets of the tricuspid valve. The tricuspid valve, one of the heart’s four valves, regulates blood flow from the right atrium to the right ventricle, preventing backflow between these chambers.

2. Abbott Spinal Cord Stimulation (SCS) Systems
Manufacturer/developer: Abbott Medical
Date of FDA approval: May 30, 2024
Approved for: Chronic, hard-to-manage pain in the torso, arms and legs.
Abbott’s SCS devices deliver its proprietary BurstDR stimulation waveform, a superior therapy designed to more closely mimic how pain signals travel to the brain and treat the emotional suffering related to pain. The SCS system comprises an implantable pulse generator (IPG), lead wires, an external clinician programmer and a patient controller. Spinal cord stimulators provide gentle electrical stimulation to the spinal cord to alleviate chronic, intractable pain. The signal generator, implanted near the hip, sends electrical pulses to the spinal cord through electrodes attached to lead wires positioned near the spinal cord.
3. Agent Paclitaxel-Coated Balloon Catheter
Manufacturer/developer: Boston Scientific Corporation
Date of FDA approval: February 29, 2024
Approved for: It is used to treat arteries that supply blood to the heart that have narrowed or blocked again after being treated with a stent.

The Agent paclitaxel-coated balloon catheter (Agent DCB) is a device that uses a thin tube with a balloon at the tip to reopen arteries supplying blood to the heart that have become blocked or narrowed due to coronary artery disease (CAD). The balloon’s outer surface is coated with the drug paclitaxel, a safe and effective measure to prevent the arteries from narrowing again.
4. Liaison Biotrin Parvovirus B19 IgG Plus
Full name of the medical technology: Liaison Biotrin Parvovirus B19 IgG Plus, Liaison Biotrin Control Parvovirus B19 IgG Plus
Manufacturer/developer: DiaSorin Inc.
Date of FDA approval: March 29, 2024
Approved for: To detect exposure to human parvovirus B19.
For this lab test, a blood sample is drawn from the patient and sent to a clinical laboratory. The method for qualitative detection of specific IgG to parvovirus B19 is an indirect sandwich chemiluminescence immunoassay, a crucial step in diagnosing and treating patients. The liquid portion of the blood (serum or plasma) containing antibodies is separated and tested using the Liason Biotrin parvovirus B19 IgG plus test on a Liason XL analyzer. In this process, the sample is mixed with specific test chemicals. If parvovirus B19 antibodies are present, they bind to the chemicals and particular particles in the test, producing a light signal. The Liason XL analyzer then measures this light signal, providing vital information for patient care.
5. Vercise PC, Vercise Gevia and Vercise Genus Deep Brain Stimulation (DBS) Systems
Manufacturer/developer: Boston Scientific Corporation
Date of FDA approval: February 29, 2024
Approved for: The Vercise PC, Vercise Gevia and Vercise Genus DBS systems serve as an additional treatment option to help alleviate certain symptoms or tremors linked to essential tremor. They are intended for individuals whose condition is not sufficiently managed with medication and whose tremors significantly impact their ability to perform daily activities.
DBS systems are implantable devices that deliver low-intensity electrical pulses to nerve centers in the brain. These DBS systems consist of the following components: An IPG (non-rechargeable or rechargeable); a clinician programmer (a program on a tablet that provides healthcare provider information); a patient controller; and a lead kit and extension kit. The pulse generator implanted in the body sends electrical pulses through wires (leads) to a specific part of the brain, the ventral intermediate nucleus of the thalamus (VIM), to reduce tremors in the arms and hands. The electrical current’s intensity and frequency can be managed with the patient controller.
6. Intellanav Stablepoint Ablation Catheter
Full name of the medical technology: Intellanav Stablepoint Ablation Catheter & Force Sensing System on the Rhythmia HDX Mapping System
Manufacturer/developer: Boston Scientific Corporation
Date of FDA approval: February 26, 2024
Approved for: It can treat adults with drug-resistant, recurrent, symptomatic paroxysmal atrial fibrillation (AF).
The Intellanav Stablepoint ablation catheter, a non-invasive solution, uses radiofrequency (RF) energy delivered through its tip to destroy heart tissue cells responsible for causing arrhythmias or irregular heart rhythms. A doctor inserts the catheter through a small incision in a vein in the groin and guides it to the heart. The catheter’s tip is positioned at various locations within the heart, where the doctor uses a generator to deliver RF energy. This energy heats and scars specific heart tissue cells, disrupting the electrical signals that cause abnormal heart rhythms. Once the treatment is complete, the catheter is removed.
7. Xstim Spine Fusion Stimulator
Manufacturer/developer: Xstim Inc.
Date of FDA approval: February 9, 2024
Approved for: It is indicated as an additional electrical treatment after lumbar spinal fusion surgery for one or two levels of the spine at home all day for nine months. This device is available by prescription and intended for adult patients only.

The Xstim spine fusion stimulator, a bone growth stimulator (BGS) designed for the lower back, is a powerful tool for post-operative care. Following a spinal fusion procedure, the Xstim spine fusion stimulator electrodes are positioned on the lower back, with one electrode on each side of the fusion site. Once the device is turned on and the electrodes are detected, it enters “Treatment Mode.” In this mode, the device automatically delivers a 60 kHz sine wave between the electrodes, creating a low-energy electrical field at the fusion site to support and promote spinal fusion.
8. Xact Carotid Stent System
Manufacturer/developer: Abbott Vascular Inc.
Date of FDA approval: February 7, 2024
Approved for: This device is intended to help widen the carotid arteries in patients who are at high risk for complications from traditional surgery (carotid endarterectomy). It is recommended for those who need a minimally invasive procedure, such as carotid angioplasty and stenting, to treat blocked arteries, based on their doctor’s evaluation.
The Xact carotid stent system is used to reopen narrow parts of the carotid arteries in the neck that supply blood to the brain. The system comprises a delivery catheter and a self-expanding metal stent made of nitinol. Initially, the Enroute transcarotid neuroprotection system (Enroute NPS) is inserted through tubes placed in the patient’s leg and neck, with the tubes connected by a flow controller that manages the flow of liquids and gases. It redirects blood flow at the treatment site in the neck, and once it is positioned, the doctor inserts the Xact delivery catheter into the neck and guides the Xact carotid stent to the blocked area of the carotid artery. The stent is then released from the catheter, expanding automatically to open the blood vessel over the blockage. The stent delivery catheter and the Enroute NPS are then removed, leaving the stent permanently in place to support the newly opened section of the artery.
9. Spectra WaveWriter, WaveWriter Alpha, WaveWriter Alpha Prime SCS Systems
Manufacturer/developer: Boston Scientific Corporation
Date of FDA approval: February 5, 2024
Approved for: WaveWriter SCS systems are used to help manage chronic, intractable pain in the trunk, arms or legs, including one-sided or two-sided pain associated with the various conditions.
The Spectra WaveWriter SCS system is the first SCS system designed to personalize pain relief through the power of combination therapy and the simplicity of waveform automation. The system is designed with user-friendliness in mind, with central parts including an implanted pulse (signal) generator that is connected to one or two implanted leads, a remote control and a clinician programmer (a program on a tablet that provides healthcare provider information). The IPG receives radio signals from the controller used by the patient. The signals tell the IPG when to deliver the right stimulation (activation) to the spinal cord.
10. Evoque Tricuspid Valve Replacement System
Manufacturer/developer: Edwards Lifesciences LLC
Date of FDA approval: February 1, 2024
Approved for: It is intended to treat patients with a severely leaky tricuspid valve (patients with symptomatic severe TR despite optimal medical therapy) often caused by an enlarged heart or damaged native valve flaps (called leaflets).
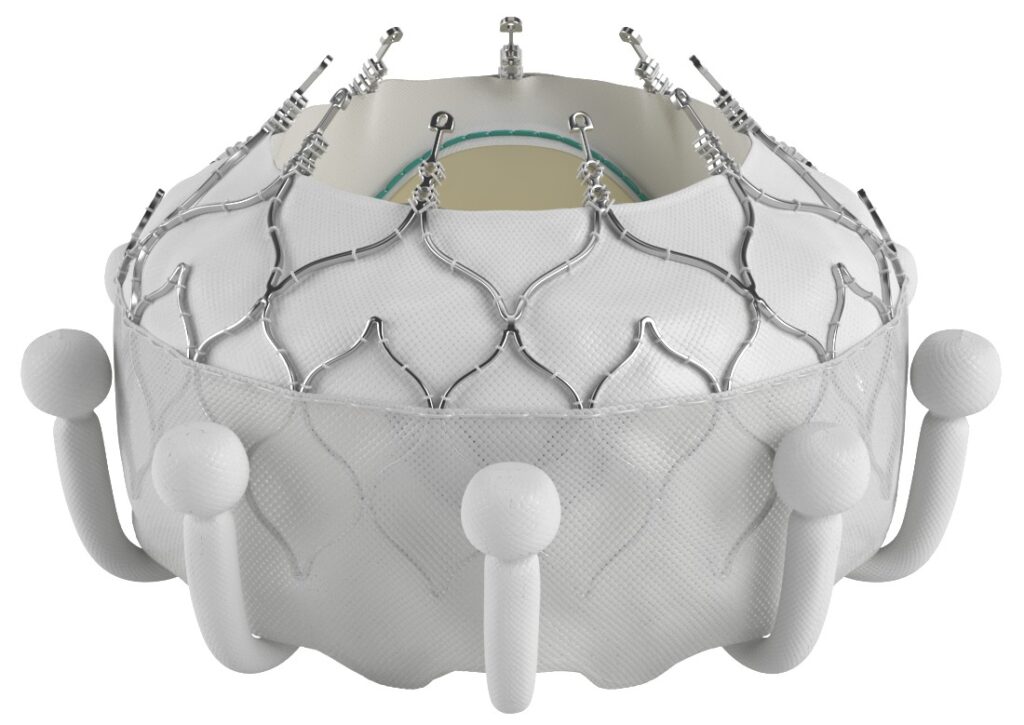
The Evoque tricuspid valve replacement system consists of an artificial tricuspid valve (Evoque) and a delivery catheter. It offers a less invasive alternative to open-heart surgery for replacing a damaged tricuspid valve. The tricuspid valve plays a crucial role in preventing blood from flowing backward into the right atrium from the right ventricle. In this procedure, the doctor first compresses the Evoque valve and attaches it to the end of the delivery catheter. The catheter is then inserted through a large vein in the groin (femoral vein) and carefully guided through the blood vessels to the heart. Once it reaches the diseased tricuspid valve, the doctor releases the new valve, allowing it to expand and securely anchor itself in place. Finally, the catheter is removed, leaving the new valve to take over the function of the original one. The new valve opens and closes to ensure proper blood flow in the heart, helping it work more effectively.
11. TZ Medical Multi-Function Defibrillation Electrodes and Adaptors
Full name of the medical technology: TZ Medical Adult Multi-Function Defibrillation Electrodes and Adapters and TZ Medical Pediatric Multi-Function Defibrillation Electrodes and Adapters
Manufacturer/developer: TZ Medical, Inc
Date of FDA approval: January 9, 2024
Approved for: The TZ Medical multi-function defibrillation electrodes are indicated for use by trained first responders with compatible defibrillators to treat abnormal heart rhythms, including ventricular fibrillation (VF), ventricular tachycardia (VT) and bradycardia (heart beats too slow).
The TZ Medical multi-function defibrillation electrodes adhere to a person’s chest and are connected to a compatible defibrillator directly or via an adaptor. Once connected, the defibrillator monitors the person’s heart rhythm to detect and address life-threatening arrhythmias, delivering an electrical pulse or shock through the electrodes as needed.
12. Minima Stent System
Manufacturer/developer: Renata Medical
Date of FDA approval: August 28, 2024
Approved for: Coarctation of the aorta and pulmonary artery stenosis in neonates, infants and children weighing at least 3.3 pounds.
For young patients facing severe artery narrowing, the Minima stent system offers a lifesaving solution with its expandable cobalt-chromium mesh tube. Delivered through a minimally invasive catheter system, the stent is positioned using X-ray imaging, then expanded via an inflatable balloon. Once in place, it acts as a scaffold, restoring proper blood flow. In a clinical trial, participants demonstrated a 98% success rate after six months post implantation, with arteries widened successfully and no stent fractures observed. The very first patient successfully received the Minima stent later last year.
13. Altius Direct Electrical Nerve Stimulation System
Manufacturer/developer: Neuros Medical, Inc.
Date of FDA approval: August 26, 2024
Approved for: Reducing chronic phantom limb pain and residual limb pain in the legs of adult amputees post-amputation.
Phantom limb pain can feel like a cruel echo, persisting even after the loss of a limb. The Altius direct electrical nerve stimulation system provides a new way to alleviate this debilitating pain. This implantable device uses a cuff electrode wrapped around the target nerve in the amputated leg. By delivering high-frequency alternating current (HFAC) signals via an implanted pulse generator, it disrupts pain signals, offering patients significant relief. Over 12 months, patients saw days with pain reduced by 50%, an 81% decrease or elimination of opioid use and 45% of patients had a better quality of life.
14. Shield
Manufacturer/developer: Guardant Health, Inc.
Date of FDA approval: July 26, 2024
Approved for: Screening adults aged 45 or older with an average risk for colorectal cancer.

The Shield test provides a non-invasive approach to colorectal cancer screening by analyzing blood for key DNA changes linked to cancer. After a simple blood draw using the Guardant Shield blood collection kit, the sample is sent to a specialized lab. There, plasma is separated, and tumor DNA is identified by detecting genetic mutations, methylation patterns and fragmentation signals. A clinical study of 7,861 participants found the Shield test detected colorectal cancer in 83% of cases, highlighting its value for early diagnosis. Positive results prompt further diagnostic colonoscopy, while negative results support continued routine screening.
15. Unipure C3F8 Ophthalmic Gas, Unifeye Gas Delivery System, Unipexy Gas Delivery System
Manufacturer/developer: Alcon Research, LLC
Date of FDA approval: July 2, 2024
Approved for: Treating uncomplicated retinal detachments.
The Unipure C3F8 ophthalmic gas, delivered via the Unifeye or Unipexy gas delivery systems, serves as a minimally invasive option for treating retinal detachment. Injected into the vitreous cavity — the gel-like interior of the eye — it forms a gas bubble that gently pushes the retina back into place, preventing fluid from entering the retinal tear. This temporary scaffold holds the retina stable while healing occurs, with the gas naturally dissolving over six to eight weeks. Literature review data from similar gas showed an 81.9% reattachment success rate at three months, supporting its potential in vision restoration.
16. Mynx Control Venous Vascular Closure Device (VCD)
Full name of the medical technology: Mynx Control Venous Vascular Closure Device (VCD) 6F-12F
Manufacturer/developer: Cordis US Corporation
Date of FDA approval: June 27, 2024
Approved for: Closing femoral vein puncture sites after catheterization procedures.

The Mynx Control venous VCD is designed to streamline the closure of puncture sites in the femoral vein following catheterization. Using a delivery system, it places an absorbent gel at the access site, which expands upon contact with blood to seal the vessel walls. In a trial with 100 patients, the device demonstrated no major complications, an average bleeding stop time of 2 minutes, with patients walking within approximately 2.5 hours.
17. Edwards Sapien 3, Sapien 3 Ultra and Sapien 3 Ultra Resilia Transcatheter Heart Valve (THV) System
Manufacturer/developer: Edwards Lifesciences LLC
Date of FDA approval: May 23, 2024
Approved for: Replacing a failing, previously implanted surgical biological mitral valve in patients at intermediate or higher risk from open-heart surgery.
The Sapien 3 THV system expands treatment options for patients with failing mitral tissue valves who face significant risks with open-heart surgery. The device, made of cow tissue mounted on a cobalt-chromium frame, is delivered through a balloon catheter inserted via the femoral vein. Once guided to the failing valve, it is expanded to restore proper blood flow. Clinical data from 499 patients confirmed its safety and effectiveness, providing a less invasive alternative to improve heart function and reduce life-threatening complications. Further, real-world data from more than 9,000 patients demonstrated low mortality rates, larger effective valve openings and no significant leakage around the valve in 84.4% of cases.
18. BD Onclarity HPV Assay
Manufacturer/developer: Becton, Dickinson and Company (BD)
Date of FDA approval: May 14, 2024
Approved for: Detecting high-risk human papillomavirus (HPV) types associated with cervical cancer.
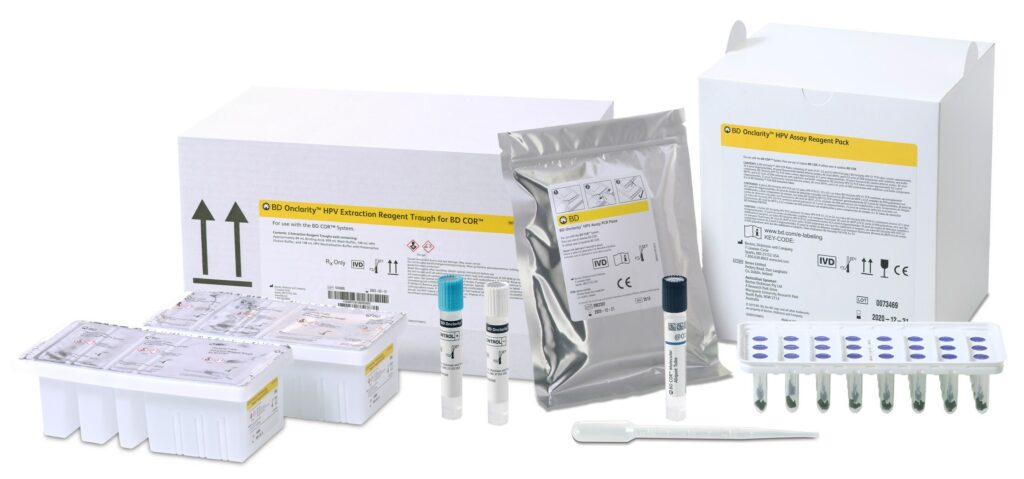
The BD Onclarity HPV assay enhances cervical cancer screening by identifying DNA from 14 high-risk HPV types. Using cervical or self-collected vaginal specimens, the test works with the BD Viper LT and BD COR systems to detect individual genotypes, such as HPV 16, 18 and 31, and grouped results for other types. Negative results help rule out cancer-causing infections, while positive results guide follow-up procedures. By helping rule out cancer-causing infections or identify those requiring further attention, the assay aids healthcare providers in better managing cervical cancer risk.
19. cobas HPV Test for 4800 System
Manufacturer/developer: Roche Molecular Systems, Inc.
Date of FDA approval: May 14, 2024
Approved for: Detecting high-risk HPV in cervical and self-collected vaginal specimens for cervical cancer screening.
The cobas HPV test is a laboratory diagnostic designed to identify 14 high-risk HPV types associated with cervical cancer. Used on the cobas 4800 system, it processes both clinician-collected cervical samples and self-collected vaginal specimens, making it a versatile tool for screening. One clinical study confirmed the cobas HPV test’s safety and effectiveness for routine cervical cancer screening, while a second, validated its reliable performance with self-collected vaginal samples compared to clinician-collected cervical samples.
20. cobas HPV for 5800/6800/8800 Systems
Manufacturer/developer: Roche Molecular Systems, Inc.
Date of FDA approval: May 14, 2024
Approved for: Detection of HPV in cervical samples (specimens) and self-collected vaginal samples.
Roche’s cobas HPV test is a self-collection solution, one of the first in the US, according to Roche. The automated diagnostic assay, designed for use on the cobas 5800, 6800 and 8800 systems, detects HPV in cervical samples and self-collected vaginal specimens. Individuals collect their own vaginal sample in a clinical setting, which is then sent to a laboratory for testing using Roche’s cobas molecular instrument. It detects the presence of DNA of high-risk HPV (hrHPV) — strains that cause precancerous cell changes and are the main cause of more than 90% of cervical cancers — in samples (liquid-based cytology [LBC] specimens) using real-time PCR. The test offers rapid results, generating up to 96 results in about 3 hours, 384 results for the cobas 6800 system and 1,056 results for the cobas 8800 system in an 8-hour period. If the result is positive for HPV, the individual can follow up with a healthcare provider for further evaluation and care. HPV self-collection provides a convenient and accessible screening method.
21. ColoSense
Manufacturer/developer: Geneoscopy, Inc.
Date of FDA approval: May 3, 2024
Approved for: Colorectal cancer screening test for individuals 45 years of age or older.
ColoSense is a laboratory test that detects eight different RNA biomarkers and trace amounts of hemoglobin (as a measure of blood) in human stool that is not visible to the naked eye (fecal occult blood). The test collection kit includes a container for stool collection and a separate sampler for hemoglobin testing, requiring only a single stool sample to collect both. No bowel preparation, dietary changes or medication restrictions are needed to complete the test. According to Geneoscopy, ColoSense is the first non-invasive colorectal cancer screening test “to provide a dynamic view of disease activity by using RNA biomarkers.” A positive or negative result is determined based on the combined analysis of selected RNA biomarkers, hemoglobin levels and smoking status. ColoSense results are then provided to healthcare providers for additional evaluation.
22. nAbCyte Anti-AAVRh74var HB-FE assay
Manufacturer/developer: LabCorp Drug Development
Date of FDA approval: April 25, 2024
Approved for: Laboratory test for adults with moderate or severe hemophilia B. Companion diagnostic for Pfizer’s hemophilia B gene therapy Beqvez (idanacogene elaparvovec-dzkt).
The test is designed for identifying individuals with moderate to severe hemophilia B (congenital factor IX [FIX] deficiency) who may benefit from the gene therapy Beqvez. The genetic bleeding disorder is caused by the absence of blood clotting factor IX. The companion diagnostic, which was approved alongside Beqvez in April 2024, detects the presence of neutralizing antibodies against the adeno-associated virus vector AAVRh74var, which is used in Beqvez to deliver a functional copy of the FIX gene. The test is used to determine whether a patient has previously been infected with the virus. If the test detects AAVRh74var antibodies, the individual is considered AAVRh74var antibody positive and is not eligible for treatment with Beqvez. Conversely, if no AAVRh74var antibodies are detected, the individual is presumed to be AAVRh74var antibody negative and may be a candidate for treatment with Beqvez. For the test, blood samples are sent to Labcorp-Monogram Biosciences Laboratories for analysis from which the serum is tested for the presence of neutralizing antibodies against the virus.
23. Esprit BTK Everolimus Eluting Resorbable Scaffold System
Manufacturer/developer: Abbott Medical
Date of FDA approval: April 26, 2024
Approved for: Chronic limb-threatening ischemia (CTLI).
Esprit BTK (below-the-knee) is a bioabsorbable, drug-coated mesh implant designed to expand a narrowed artery in the lower leg, improving blood flow to the leg and foot to address ischemia. The scaffold is placed inside the lower leg artery to help keep it open while gradually releasing a drug to reduce the risk of re-narrowing. To implant the scaffold, a physician inserts a catheter into a blood vessel that has the scaffold mounted on a small balloon at its tip, guiding it to the affected artery. Once positioned at the narrowed section, the balloon is inflated, expanding the scaffold and pressing it against the artery wall. Additional balloon inflations within the scaffold may be performed to ensure optimal expansion. Abbott says the Esprit BTK system is the only BTK device that simultaneously delivers drug and provides biomechanical support to the leg.
24. Lumicell Direct Visualization System (DVS)
Manufacturer/developer: Lumicell, Inc.
Date of FDA approval: April 17, 2024
Approved for: Fluorescence imaging in adults with breast cancer as an adjunct for the intraoperative detection of residual cancer cells/tissue within the resection cavity after removal of the primary specimen during lumpectomy surgery.
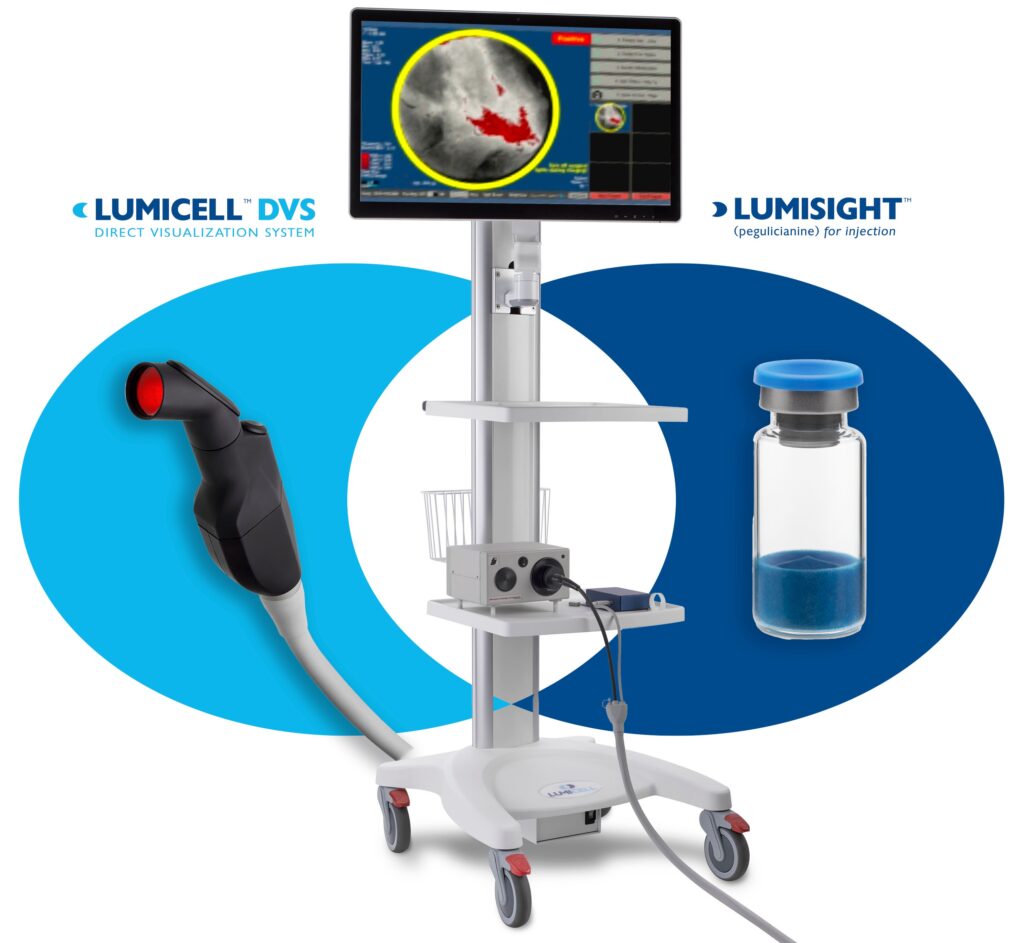
The Lumicell DVS is a drug-device combination product (DDCP) that provides fluorescence imaging for adjunct breast cancer tissue detection after lumpectomy surgery. The system includes an optical imaging agent, Lumisight (pegulicianine), that is administered intravenously 2 to 6 hours prior to imaging, along with a fluorescence imaging device. The device excites the Lumisight agent within the cavity and captures real-time fluorescence images, which can help identify potential residual cancer tissue. According to Lumicell, the product, which has a diagnostic accuracy of 84%, enables surgeons to perform real-time scanning of the breast cavity after a lumpectomy to identify and remove any remaining cancerous tissue that might have been overlooked, potentially reducing the need for additional surgeries.
25. Gore Excluder Conformable AAA Endoprosthesis (EXCC)
Manufacturer/developer: W.L. Gore & Associates, Inc.
Date of FDA approval: April 5, 2024
Approved for: Intended for use in patients with an infrarenal abdominal aortic aneurysm whose anatomy meets the criteria specified in the instructions for use.
The Gore Excluder conformable AAA endoprosthesis is a stent graft designed to treat an aneurysm — a weakened, bulging section — of the infrarenal abdominal aorta, located below the renal (kidney) arteries. The multi-component system includes stent grafts, which are flexible fabric tubes supported by a metal framework that remain permanently implanted, housed in delivery catheters. The device is inserted into the femoral artery (a major blood vessel in the upper leg) through a small incision, and then carefully guided to the aneurysm site in the abdominal aorta. Once positioned, the stent graft creates a new pathway for blood flow, reducing pressure on the aneurysm and potentially preventing its growth or rupture, which could be life-threatening. Gore Medical says the Gore Excluder conformable AAA endoprosthesis offers the only on-label endovascular aneurysm repair (EVAR) solution in highly angulated aortic necks up to 90 degrees and neck length of 10 mm. The conformable stent graft and delivery system builds on the company’s Gore Excluder device family.
26. TruSight Oncology Comprehensive
Manufacturer/developer: Illumina, Inc
Date of FDA approval: August 21, 2024
Approved for: The laboratory test serves as a companion diagnostic to identify individuals who are likely to benefit from a particular drug or therapy.
The TruSight Oncology Comprehensive in vitro diagnostic (IVD) is a cancer biomarker test that profiles a patient’s solid tumor to help identify certain immuno-oncology or clinically actionable biomarker(s) that will help identify patients suited for treatment with a certain targeted therapy or enrollment in a clinical trial. For instance, individuals with solid malignant tumors showing specific RNA alterations in the NTRK1, NTRK2 or NTRK3 genes, as detected by the test, may be candidates for personalized treatment with Bayer’s Vitrakvi (larotrectinib). Similarly, people with non-small cell lung cancer (NSCLC) who have specific RNA mutations in the RET gene may benefit from personalized therapy with Eli Lilly’s Retevmo (selpercatinib). The diagnostic tests for more than 500 genes to profile tumors. Illumina says TruSight Oncology Comprehensive is “the first US FDA-approved, distributable comprehensive genomic profiling IVD kit with pan-cancer companion diagnostic claims.”
27. Simplera system
Manufacturer/developer: Medtronic MiniMed, Inc.
Date of FDA approval: July 24, 2024
Approved for: Continuous glucose monitoring (CGM) system for individuals with type 1 or type 2 diabetes.
The Simplera CGM system provides real-time blood sugar (glucose) values through a sensor and an app that work together to help people to manage their diabetes. The Simplera sensor measures glucose levels in the interstitial fluid of the arm and wirelessly sends the data to the app. The Simplera app, compatible with devices like smartphones and tablets, displays glucose levels, trends and provides alerts for low or high glucose values based on user-configured settings. Users can also manually log details such as blood glucose readings, meals, exercise or insulin doses. The device is half the size of Medtronic’s previous CGMs. Medtronic says the new smaller and simpler design makes the insertion and wear experience simpler and easier, eliminating the need for overtape.
28. Unipure SF6 Ophthalmic Gas in the Unifeye Gas Delivery System and Unipure SF6 Ophthalmic Gas in the Unipexy Gas Delivery System
Manufacturer/developer: Alcon Research, LLC
Date of FDA approval: August 26, 2024
Approved for: The Unipure SF6 ophthalmic gas is a gas that can be injected into the eye for the treatment of uncomplicated retinal detachments.
The Unifeye gas delivery system injects filtered Unipure SF6 ophthalmic gas mixed with filtered air into the vitreous cavity, the gel-like space inside the eye. Once injected, the gas forms a bubble that aids in reattaching the retina by pressing the detached retina back into place. This prevents additional fluid from entering the vitreous cavity and passing behind the detached retina through the retinal tear. As the retinal tear or hole (retinal break) heals, the gas bubble blocks fluid from accumulating between the detached retina and its supporting structures. The bubble stabilizes the retina until it gradually dissolves, typically within 12 to 14 days.
29. Oncomine Dx Target Test
Manufacturer/developer: Life Technologies Corporation
Date of FDA approval: October 17, 2024
Approved for: Detecting IDH1 and IDH2 mutations in astrocytoma and oligodendroglioma tumors to guide personalized treatment with Voranigo (vorasidenib).
The Oncomine Dx target test is an FDA-approved companion diagnostic (CDx) designed to identify genetic changes in tumor tissue, enabling personalized treatment decisions. For patients with astrocytoma and oligodendroglioma, the test detects mutations in the IDH1 and IDH2 genes — key markers for eligibility to receive Voranigo, the first FDA-approved targeted therapy for grade 2 IDH-mutant glioma. Using a small sample of tumor tissue embedded in wax and sliced thinly, the test isolates DNA and RNA, which are then analyzed with chemical reagents to detect mutations in the IDH1 and IDH2 genes. These results are sent to the physician, providing critical genetic insights to guide treatment decisions.
30. PyloPlus Urea Breath Test (UBT) System
Manufacturer/developer: ARJ Medical Inc
Date of FDA approval: January 11, 2024
Approved for: Detecting Helicobacter pylori (H. pylori) infections in individuals aged three years and older.
The PyloPlus UBT system offers a simple and non-invasive approach to diagnosing H. pylori infections, a common cause of stomach ulcers. The process begins with the patient providing a baseline breath sample, followed by ingestion of a safe drug mixture containing radioactive carbon. Twenty minutes later, a second breath sample is collected and analyzed for elevated radioactive carbon dioxide levels, which indicate the presence of H. pylori. Clinical studies have validated its use in both adult and pediatric populations, making it a versatile diagnostic tool for managing gastrointestinal health.









Join or login to leave a comment
JOIN LOGIN