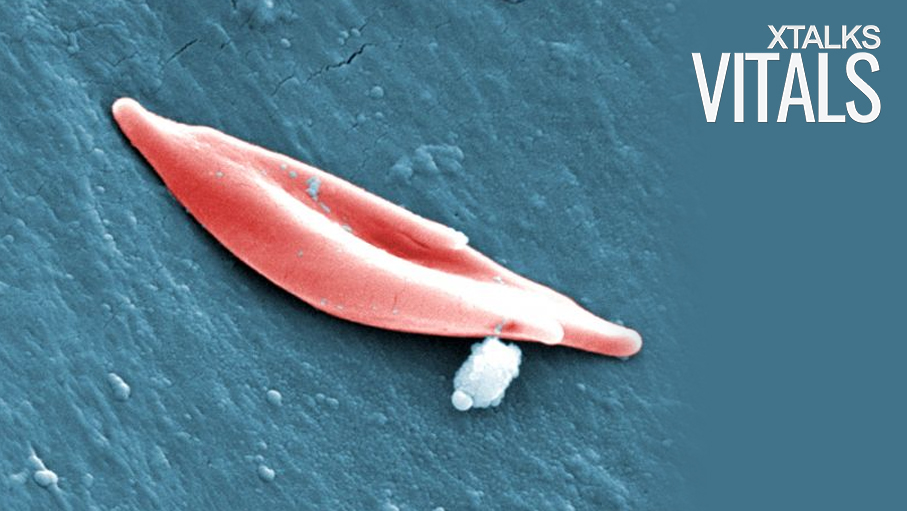
Researchers at St. Jude’s Children’s Research Hospital have pioneered the use of CRISPR gene editing as a therapeutic for sickle cell disease and other blood disorders. The international, proof-of-principal study was published in the journal, Nature Medicine, and could offer new hope to those with sickle cell disease and beta-thalassemia.
“Our approach to gene editing is informed by the known benefits of hereditary persistence of fetal hemoglobin,” said Dr. Mitchell J. Weiss, chair of the St. Jude Department of Hematology and the senior author on the study. “It has been known for some time that individuals with genetic mutations that persistently elevate fetal hemoglobin are resistant to the symptoms of sickle cell disease and beta-thalassemia, genetic forms of severe anemia that are common in many regions of the world. We have found a way to use CRISPR gene editing to produce similar benefits.”
While both fetal and adult forms of hemoglobin are both responsible for carrying oxygen in red blood cells, they differ based on their molecular makeup of four subunits. Adult hemoglobin contains two beta subunits, whereas fetal hemoglobin consists of gamma subunits instead.
As sickle cell disease and beta-thalassemia are both caused by mutations in the gene encoding the beta subunit, the symptoms of disease slowly begin to manifest themselves as the adult hemoglobin levels increase. Mutations in the beta subunit can impair the oxygen carrying capacity of adult hemoglobin, leading to red blood cell death and hypoxia in vital organs and tissues.
Since sickle cell mutations do not have an impact on fetal hemoglobin – due to the lack of beta subunits – this form of hemoglobin has the potential to effectively carry oxygen in adults. By applying CRISPR gene editing, the researchers sought to prevent the gamma-to-beta switch of hemoglobin in adults, thereby reducing the symptoms associated with sickle cell disease and beta-thalassemia.
“Our work has identified a potential DNA target for genome editing–mediated therapy and offers proof-of-principle for a possible approach to treat sickle cell and beta-thalassemia,” said Weiss. “We have been able to snip that DNA target using CRISPR, remove a short segment in a ‘control section’ of DNA that stimulates gamma-to-beta switching, and join the ends back up to produce sustained elevation of fetal hemoglobin levels in adult red blood cells.”
When applying the CRISPR technique to the DNA of hematopoietic stem cells collected from patients with sickle cell disease, they were successful in stimulating those cells to express healthy fetal hemoglobin. In addition to other gene editing methods to treating sickle cell disease, this research represents a promising approach for treating the blood disorder.
“Our results represent an additional approach to these existing innovative strategies and compare favorably in terms of the levels of fetal hemoglobin that are produced by our experimental system,” said Weiss. What’s more, patients who carry naturally-occurring genetic mutations which prevent them from transitioning from fetal to adult hemoglobin, are usually asymptomatic.
Much like other applications of CRISPR, the current approach has the potential to trigger harmful off-target mutations. Before this technique can be used in human clinical trials, the researchers will need to further explore how to prevent these off-target effects, as well as assess the broad safety and efficacy considerations for patients with sickle cell disease.






Join or login to leave a comment
JOIN LOGIN