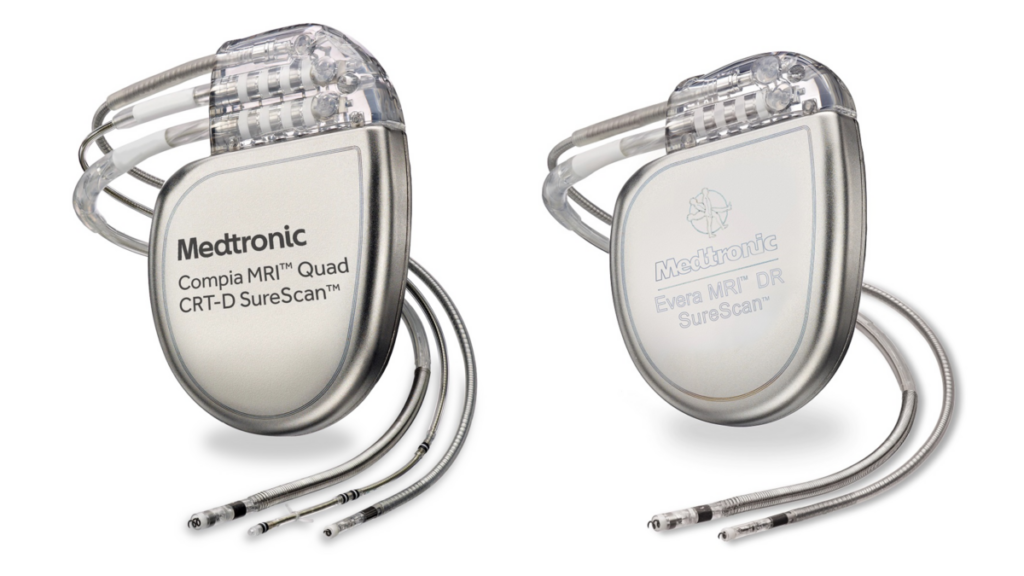
Implantable cardioverter defibrillators (ICDs) and cardiac resynchronization therapy defibrillators (CRT-Ds) are medical devices specifically designed to detect and correct irregular heartbeats or arrhythmias, such as ventricular tachycardia or ventricular fibrillation. These devices play a crucial role in providing life-saving therapy to patients who are at risk of sudden cardiac arrest. They function by generating an electric shock (cardioversion or defibrillation) that restores the heart’s normal rhythm.
Furthermore, a CRT device enhances the heart’s efficiency by sending small electrical impulses to both lower chambers of the heart. This promotes a more synchronized heartbeat, thereby improving the heart’s ability to pump blood and oxygen throughout the body.
Medtronic, a leading med tech company with a significant focus on cardiovascular devices, began notifying physicians in May 2023 about the possibility of reduced-energy or no-energy high-voltage therapy in its ICDs and CRT-Ds. To mitigate the risk associated with this issue, the company has advised physicians to reach out to patients and non-invasively reprogram the devices. In the affected population, Medtronic has documented 28 incidents, 22 injuries, but fortunately, no deaths related to this problem.
The company has issued a recall for approximately 350,000 ICDs and CRT-Ds that were manufactured post-July 2017. The US Food and Drug Administration (FDA) has classified this as a Class I recall, the agency’s most serious level of recall.
The recall affects devices manufactured with a specific (glassed) feedthrough under various brand names, which include:
- Cobalt XT, Cobalt, Crome ICDs and CRT-Ds
- Claria MRI, Amplia MRI, Compia MRI, Viva, Brava CRT-Ds
- Visia AF, Visia AF MRI, Evera, Evera MRI, Primo MRI, Mirro MRI ICDs
“Medtronic provided physicians with comprehensive patient management recommendations in the communication. We have recommended that physicians non-invasively reprogram these devices to reduce the risk for this issue,” Medtronic said in a statement on their website.
RELATED: Medtronic Expands Two Insulin Pump Recalls, Including One Over Cybersecurity Concerns
The Reasons for the Recall of Medtronic Defibrillators
Medtronic has issued a recall for all ICDs and CRT-Ds manufactured post-2017 that contain a glassed feedthrough. These devices may malfunction by delivering inadequate or no energy when high-voltage therapy is required due to the inappropriate triggering of the Short Circuit Protection (SCP) feature. The issue is more prevalent in devices equipped with a glassed feedthrough and set to deliver therapy via the AX>B pathway.
A shock of reduced energy or the lack of a shock could fail to address a life-threatening arrhythmia, potentially leading to cardiac arrest, other serious injuries or even death. Additionally, patients fitted with these devices may be exposed to further risks if they require extra surgical procedures to remove and replace the malfunctioning device.
What Does Medtronic Recommend?
Medtronic does not recommend prophylactic device replacement; instead, reprogramming devices through non-invasive routes is recommended. All high voltage therapy pathways need to be programmed to B>AX to minimize the risk for this issue. Patients with a history of high voltage therapy and Rx1 programmed AX>B should be prioritized. Patients with AX>B programming in any high voltage therapy should attend the scheduled follow-up for in-clinic device reprogramming.
When ICD and CRT-D devices in the glassed feedthrough population are programmed exclusively in the B>AX configuration, the estimated risk for a reduced- or no-energy high voltage therapy is 0.002 percent at five years and 0.005 percent at nine years. These estimates for device failure after reprogramming are consistent with historical data for device performance across all Medtronic-manufactured devices of this type and industry standards for device performance.
Medtronic recommends contacting their Technical Services or a local representative if any of the following issues are detected (as they may indicate a problem with either the device or the lead):
- Display of reduced-energy or no-energy high voltage therapy in Episode Text, irrespective of the programmed pathway.
- A consistent drop of around 50 percent in the impedance measurements of the right atrium (RA), right ventricle (RV) and left ventricle (LV) pacing leads, as this could be a precursor to potential low or non-existent energy therapy in the future.
Other Recalls by Medtronic
In June 2022, Medtronic recalled its Cobalt/Crome ICDs and CRT-Ds after receiving reports of devices with a SCP alert resulting in reduced-energy electric shock delivery instead of delivering a second phase of high voltage therapy.
In 2021, Medtronic recalled almost 240,000 defibrillators because of the potential for a rapid decrease in battery life.
In addition, in 2018, the FDA issued warning letters to Medtronic plants in Minnesota and Puerto Rico following earlier recalls of their ICDs.







Join or login to leave a comment
JOIN LOGIN