Statins are the most commonly used group of drugs to treat lipid disorders. They not only help lower cholesterol, but they are associated with anti-inflammatory and antioxidant activity. According to the Centers for Disease Prevention and Control (CDC), lipid-lowering drugs were the most common type of drug used among US and Canada adults aged 60–79.
Statins are a challenging active pharmaceutical ingredient (API) to work with, as they can assume more than two different crystalline structures (polymorphs), each of which affects its physical properties and pharmacological activity.
Consider atorvastatin calcium, one of the most widely prescribed drugs in the world. There are at least 70 known polymorphic forms of atorvastatin, but the majority of them are a thermodynamically stable crystalline form used in oral drugs. These forms tend to have low solubility, reducing their absolute bioavailability to only 14 percent. Low bioavailability is common among other statins, presenting a challenge to drugmakers to create a shelf-stable, bioavailable dosage form.
The solid form of an active pharmaceutical ingredient (API) is important to consider in the drug development process. With the right tools, scientists can determine which polymorph would be the most appropriate for their application.
To learn more about solid form selection in drug development, Xtalks spoke with Dr. Steef Boerrigter, a senior research scientist at AMRI and an expert at materials science. He has extensive experience with experimental screening technologies for polymorphs, salts and cocrystals and has developed computational methods for virtual coformer screening. He shared his insights on the importance of solubility versus stability, the different common polymorphs in pharmaceutical drug development, regulations driving solid form selection and different screening modalities in an on-demand webinar.
Xtalks: Give me an overview of why solid form selection is important in drug development.
Dr. Steef Boerrigter (SB): The energetics of the solid state affects, most importantly, the solubility behavior of drugs when they are administered orally. The release profile is determined by the dissolution rate which, in turn, is determined by the solubility of a drug. The energetics also effect the long-term stability of the drug substance. Solid form selection is finding the balance between these two aspects.
Xtalks: What are the common types of solid forms used in pharmaceutical drug development?
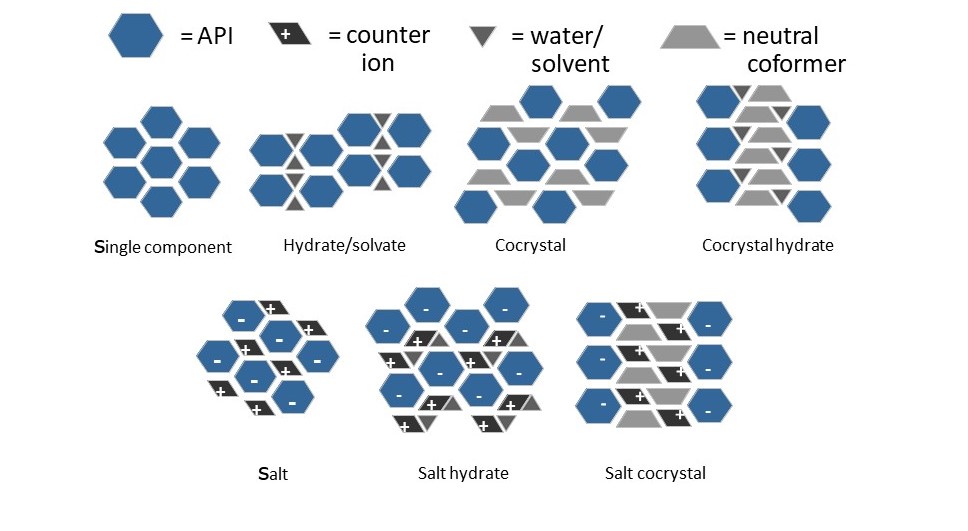
SB: The most common type of polymorphism is when we have the same molecule crystallize into different crystal lattices – a single component system. This is what we refer to as neat polymorphs or neat polymorphism. The second class is where we have different types of molecules (coformers) co-crystallizing with the active pharmaceutical ingredient (API). These are multi-component systems. When this is a solvent, we refer to that polymorph as a solvate or a solvated form. Solvates, as such, are almost never used as a drug substance because of the toxicity of solvents. However, when we replace the solvent with a non-toxic coformer, we call it a cocrystal. The use of cocrystals is becoming more and more popular.
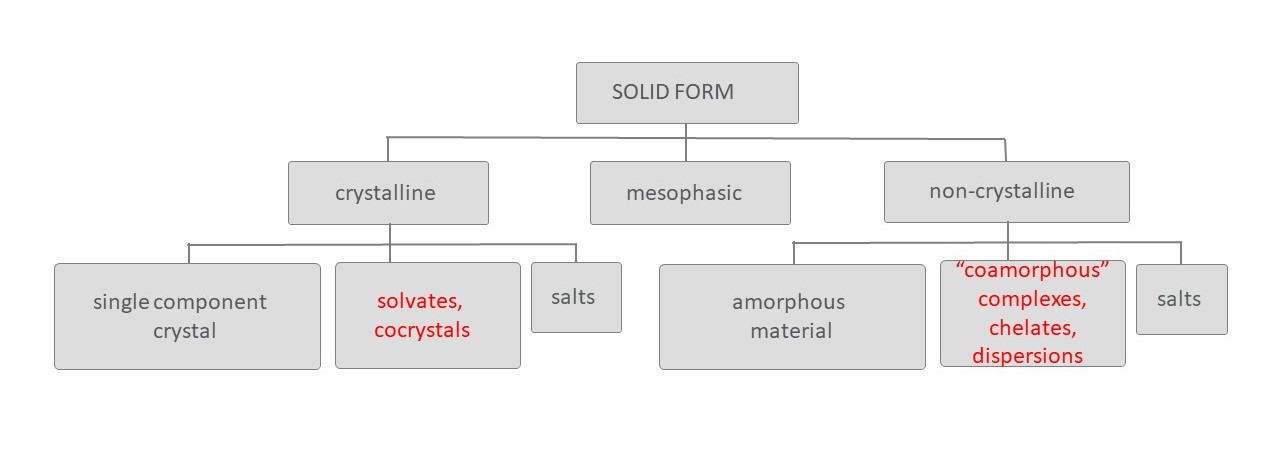
Next, we have salts, where, due to the acid-base properties of the API and coformer, the multi-component system contains ionic species. Classically, the option to pick different salt formers for the same API has been the primary handle to manipulate the solid state properties, and is still the most commonly used. Salts are typically crystalline.
Another rising class is where materials are amorphous, which is non-crystalline. Making a material amorphous can make it highly soluble but can also diminish its shelf-life. To mitigate that problem, amorphous API are typically stabilized in a dispersion system.
Xtalks: What are the different advantages, if any, of having multiple different solid forms of one API?
SB: A drug substance may have stable neat polymorphs or it may not. When we have multiple competing polymorphs, it may actually be regarded as an inconvenience rather than an advantage because you need to make sure that you don’t accidentally make the wrong polymorph during production. At the same time, it may also be an opportunity to play around with some of the properties. For instance, one polymorph may be less hygroscopic than another; one polymorph may grow as needles causing filtration problems, whereas another may not have that problem, and so on. Then there is the solubility advantage I mentioned before, although it must be said that the difference is often minimal. When a great boost in the dissolution rate is needed, it is advantageous to be able to use cocrystal or dispersion technology.
Xtalks: When do you think is the best time for pharmaceutical companies to begin integrating these aspects of solid form selection in drug development?
SB: I think the best time is at the beginning of phase one, or potentially even earlier. You may want to start thinking about this when you begin toxicity studies so that you can determine the magnitude of difference in toxicity between different levels of solubility. That way, when you test your first human exposures, you have a general idea if the form that you’re administering is going to be representative of your final drug product.
Every client is different. Some companies are required to consider solid form in early phases while others are not. Regardless, it’s a very good idea to have at least an early understanding of how complicated or how simple your form landscape is, and whether or not you’re going to have to start thinking about methods to enhance the solubility later on.
Many clients will do multiple polymorph screens as they go through development. Earlier on in the process, it may be an enabling form screen and some preliminary stability assessments. Later, we may screen for potential polymorphs due to certain solvent systems and conditions used in production. Shortly before launch we may do an intellectual property screen to ensure that all the bases have been covered.
Xtalks: How does solid form selection impact manufacturing?
SB: This is mostly important in the chemistry manufacturing and control parts of a drug filing. What the FDA wants to see is that you are producing a consistent material; that is, making a drug that has consistent performance. For the solid form, we mostly have to look at the polymorphism of the compound. We typically have an X-ray powder diffraction type of test in place to make sure that we have the polymorph consistent with the reference batch. As long as we are producing the same polymorph and also guarantee that the particle size distribution is very similar to the reference batch, then we are confident that we will have a consistent drug release profile. Depending on whether this is a critical performance attribute, this type of testing may become an FDA requirement for release of the material.
Xtalks: How does a solid form selection play a role in the intellectual property (IP) of pharmaceuticals?
SB: When companies file patents for a composition of matter, they basically lock in a molecule. They can widen their patent by effectively filing a wider range of molecules that represent the same pharmacophore. These patents are typically filed very early on in development, many years before the drug is launched. A couple of years later, that same company typically files their solid state patents. In that scenario, they want to be thorough. If there are three different polymorphs, they file all three forms. Recently, the landscape became a lot wider, because cocrystals are now also considered polymorphs. It now makes sense for a company to cover cocrystals with coformers that are generally regarded as safe in their IP portfolio.
The benefit of filing for different solid forms later on is that it can extend the patent lifetime of a drug: the clock already started ticking at the chemical compound, whereas the clock started ticking later for the particular forms.
Xtalks: Why might a company switch between different polymorphs?
SB: Companies might want to switch from a neat polymorph to a cocrystal because they’re considering different types of administration or looking for ways to optimize drug performance. For instance, if you wanted to switch from a direct release type of formulation to an extended release, then it would make sense to look at a cocrystal which would give the molecule a lower solubility than the neat form. Switching polymorphs may then offer different IP coverage if those forms weren’t already patented earlier.
Xtalks: How has drug development changed in the last 20 years?
SB: Twenty years ago, if you discovered a molecule where the solubility was too low to reach the therapeutic window then that drug would basically be shelved. Today, with the different options in solid-state development, a co-crystal, or an amorphous dispersion, may increase the solubility to where that same molecule is still a viable candidate. That is really a big difference. Our scientific understanding of solid forms has matured. Proper screening changes of late-stage surprises are greatly reduced and drugs that would have been given up on 20 years ago are now more likely to make it to patients.
To learn more about different types of polymorphs, how they are regulated by the FDA and about new screening technologies for cocrystals and dispersion systems, watch AMRI’s on-demand webinar, Solid Form Selection in Drug Development.
This article was created in collaboration with the sponsoring company and the Xtalks editorial team.







Join or login to leave a comment
JOIN LOGIN