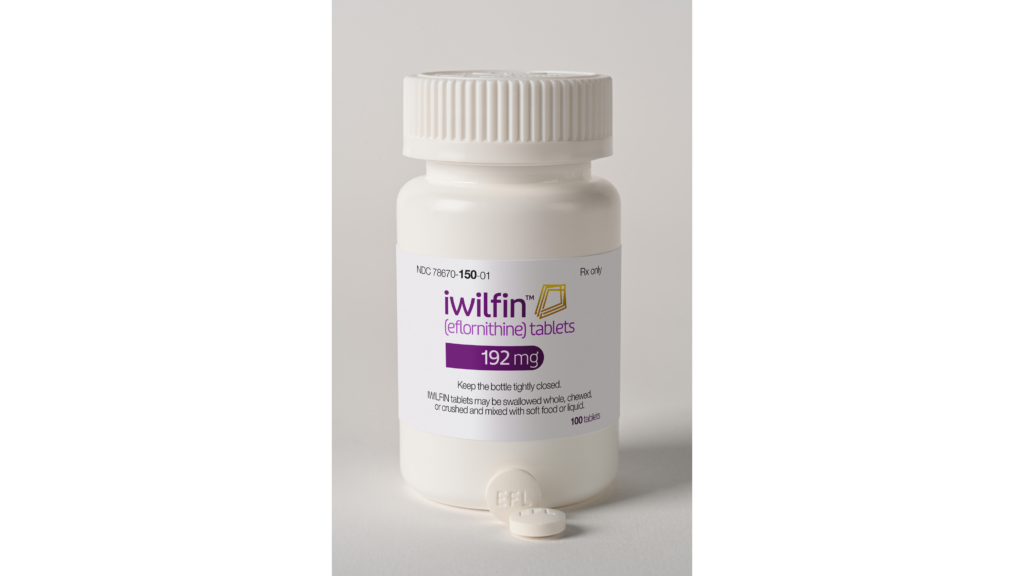
The US Food and Drug Administration (FDA) has approved Iwilfin (eflornithine) to reduce the risk of relapse in adult and pediatric patients with high-risk neuroblastoma (HRNB) who have had at least a partial response to previous multiagent, multimodality therapy including anti-GD2 immunotherapy.
This is the first FDA approval of a treatment intended to reduce the risk of relapse in pediatric patients with HRNB. Iwilfin is manufactured by US WorldMeds (USWM), LLC. It’s also the first approval for the drug in an oncology indication.
Eflornithine, an inhibitor of the enzyme ornithine decarboxylase (ODC), which is involved in cell growth and differentiation, has been around for several decades. The drug was initially developed for treating cancer, particularly cancers like colon cancer. However, its uses expanded over the years into other indications, ranging from parasitic infection to dermatology.
It was first approved by the FDA in 1989 for the treatment of African trypanosomiasis, also known as sleeping sickness, a disease caused by parasites transmitted by the tsetse fly in sub-Saharan Africa. This marked a significant advancement in the treatment of the disease, particularly for the second stage when the parasite crosses into the central nervous system. In 2000, it was approved in a topical form (under the brand name Vaniqa) for reducing excessive facial hair growth in women, a condition known as hirsutism.
The approval comes after the FDA’s Oncologic Drugs Advisory Committee (ODAC) endorsed eflornithine in a 14 to six vote in favor that sufficient evidence was provided to demonstrate the event-free survival (EFS) benefit of eflornithine to reduce the risk of relapse in pediatric patients with HRNB.
XTALKS WEBINAR: 2024’s Pharma Industry Trends and Digital Innovations
Live and On-Demand: Thursday, February 1, 2024, at 2pm EST (8pm CET/EU-Central)
Register for this free webinar to discover the forces that will impact the pharma industry in 2024 and the solutions you need.
![]()
In a statement, USWM said it partnered with the Beat Childhood Cancer Research Consortium at Penn State University, which “conducted the preclinical and clinical research to help advance this vital therapy. The Consortium represents a group of over 50 hospitals that offer collaboration through a network of childhood cancer clinical trials.”
Neuroblastoma is a type of cancer that most commonly affects young children, often occurring in infants and children under the age of five. Although a rare cancer, it is one of the more common childhood cancers. It accounts for 50 percent of all cancers in infants, which makes it the most common tumor in infants younger than one year of age. In the US, approximately 650 to 800 children are diagnosed with neuroblastoma each year.
Neuroblastoma arises from neuroblasts, which are immature nerve cells found in several areas of the body. It most frequently develops in and around the adrenal glands but can also occur in other areas of the abdomen, chest, neck and near the spine, where groups of nerve cells exist.
About half of patients diagnosed with neuroblastoma have high-risk disease (HRNB), which is classified by a combination of factors including age (children over 18 months old with neuroblastoma), those with a more advanced stage of the disease (such as stage 4) and cytogenetic characteristics such as MYCN amplification and DNA ploidy. The disease is aggressive and difficult to treat with limited treatment options.
Related: Ogsiveo Receives FDA Approval as First Therapy for Desmoid Tumors
Iwilfin’s approval was based on results from a pair of studies, Study 3b and Study ANBL0032. Study 3b consisted of two cohorts and a total of 105 eligible patients with HRNB received Iwilfin orally, twice a day, until they showed signs of cancer progression, unacceptable toxicity or for a maximum of two years. Study ANBL0032, which served as an external control arm, involved 1,241 patients and compared different treatment methods specifically in pediatric patients with HRNB.
Patients eligible for comparative analysis from both studies were matched using propensity scores. This analysis included 90 patients treated with Iwilfin and 270 patients in the control group from Study ANBL0032. Results of the analysis found significant improvements in both EFS and overall survival (OS) for patients treated with Iwilfin compared to the control group.
According to data from USWM, four years following immunotherapy, EFS in the group treated with Iwilfin was 84 percent compared to 73 percent of patients in the external control group. Moreover, 96 percent of patients who received Iwilfin were alive compared to 84 percent of external control patients, a 52 percent reduction in the risk of relapse and a 68 percent reduction in the risk of death. In additional analyses to confirm the results of the externally controlled study design, relapse risk reduction ranged from 57 percent to 41 percent and death risk reduction from 71 percent to 55 percent.
The most common adverse effects in patients treated with Iwilfin included otitis media, diarrhea, cough, sinusitis, pneumonia, upper respiratory tract infection, conjunctivitis, vomiting, fever, allergic rhinitis, decreased neutrophils, increased alanine aminotransferase, increased aspartate aminotransferase, hearing loss, skin infection and urinary tract infection.
Iwilfin was reviewed under the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission before the entire clinical application was filed, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
The application was granted priority review, breakthrough designation and orphan drug designation.








Join or login to leave a comment
JOIN LOGIN