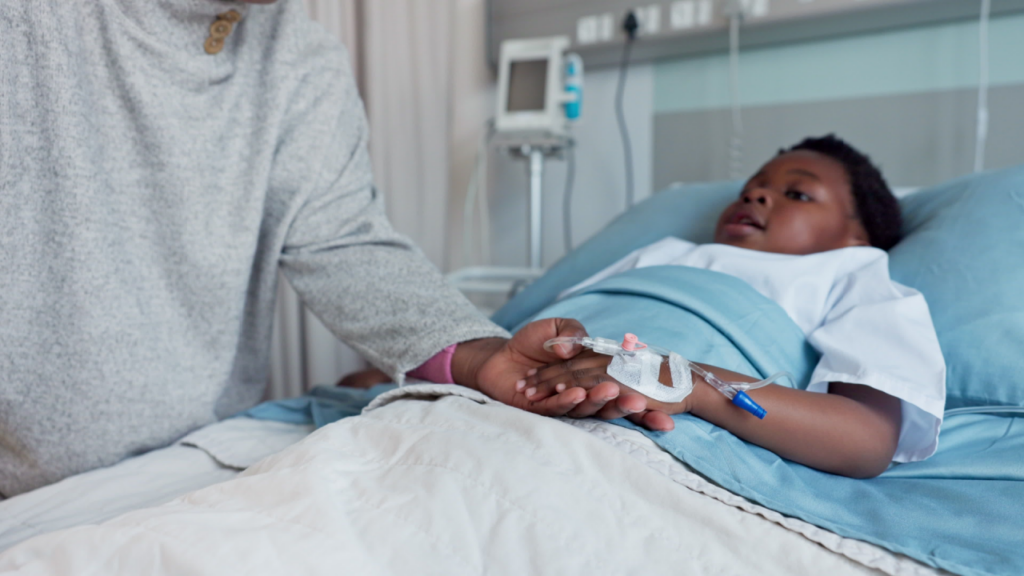
With the Casgevy FDA expansion, about 5,500 more children in the US may now be eligible for a one-time gene-editing treatment.
Sickle cell disease and transfusion-dependent beta thalassemia can affect a child’s growth, development and long-term health from an early age. Now, young children in the US have access to a one-time CRISPR-based gene-editing treatment designed to address the underlying biology of these inherited blood disorders.
Following a new FDA expansion, Casgevy (exagamglogene autotemcel) is now approved for patients as young as 2. The therapy treats children with sickle cell disease who experience recurrent pain crises caused by blocked blood flow, as well as those with transfusion-dependent beta thalassemia.
Previously, the therapy was approved only for patients aged 12 and older.
Sickle cell disease can begin causing signs in the first year of life, usually around 5 months of age, according to the CDC. Children with transfusion-dependent beta thalassemia often need regular transfusions beginning early in childhood.
According to Vertex, the decision could make about 5,500 more children in the US eligible for the one-time treatment.

Get industry leading pharma and biotech news, events and expert insights delivered to your inbox.
Sickle cell disease changes how red blood cells behave. Instead of staying flexible and round, they can become rigid and sickle-shaped. These misshapen cells can get stuck in blood vessels, blocking blood flow and triggering pain crises that often require hospital care. Over time, these blockages and chronic anemia can act like wear and tear on the body, contributing to lasting damage to organs such as the brain, lungs, kidneys and heart.
Transfusion-dependent beta thalassemia impairs the body’s ability to produce sufficient working hemoglobin, the protein that helps red blood cells carry oxygen. Children with the transfusion-dependent form often need regular blood transfusions beginning early in life. While these transfusions are necessary, the disease and iron buildup linked to frequent transfusions can eventually affect the liver, heart, bones and growth.
Casgevy is made from a patient’s own blood stem cells. The treatment process begins with collecting those cells. They are then edited outside the body using CRISPR/Cas9 technology, a gene-editing tool that can be directed to a specific part of DNA.
To make room for the treatment, patients first undergo conditioning, a high-intensity step that clears space in the bone marrow. Once the marrow is ready, the edited cells are infused back into the patient via an IV as a single dose, where they can take root and begin producing new blood cells with higher fetal hemoglobin levels.
Fetal hemoglobin is a form of hemoglobin naturally found before birth that usually declines after birth. In sickle cell disease, higher fetal hemoglobin levels can help keep red blood cells from warping into the sickle shape. In beta thalassemia, higher fetal hemoglobin and total hemoglobin levels can reduce or eliminate the need for regular red blood cell transfusions.
While the clinical trials focused on children aged 5 to 11, the FDA extended approval to children as young as 2 based on existing study results and evidence about the treatment’s characteristics and mechanism of action.
In the sickle cell disease trial, 11 children were included. All eight patients who could be evaluated for efficacy met the main study goal: no severe pain crises, as defined in the trial, for at least 12 consecutive months within the first two years after Casgevy infusion.
Results in beta thalassemia were also based on a small pediatric trial. Among 15 children enrolled, eight of the nine patients who could be evaluated for efficacy achieved transfusion independence, meaning they did not need red blood cell transfusions for at least one full year. The median duration of transfusion independence was 20.1 months.
The horizon for sickle cell care is also expanding beyond gene therapy. Novo Nordisk plans to seek regulatory approval in the second half of 2026 for etavopivat, a once-daily oral therapy that met both co-primary endpoints in a Phase III trial of patients aged 12 and older.
The FDA’s expanded approval makes Casgevy the first FDA-approved gene therapy available to children as young as 2 with sickle cell disease.
FAQs
What does “one-time therapy” mean for Casgevy?
Casgevy is given as a single infusion after a patient’s blood stem cells are collected, edited and returned to the body. The full process takes several steps, but the therapy is not taken daily or given as ongoing transfusions.
Does Casgevy cure sickle cell disease or beta thalassemia?
Casgevy is designed to address the underlying biology of these inherited blood disorders. It may provide long-term relief from severe pain crises or regular transfusions, but patients are still followed for years after treatment.
Were 2-year-olds included in the clinical trials?
The pediatric trials focused on children aged 5 to 11. The FDA extended the approval to children as young as 2 using those results and other evidence about how the treatment works in the body.
How is Casgevy different from regular blood transfusions?
Blood transfusions help manage beta thalassemia by replacing red blood cells. Casgevy uses gene editing to help a patient’s own cells produce more functional hemoglobin over time.
If you want your company to be featured on Xtalks.com, please email [email protected].






Join or login to leave a comment
JOIN LOGIN