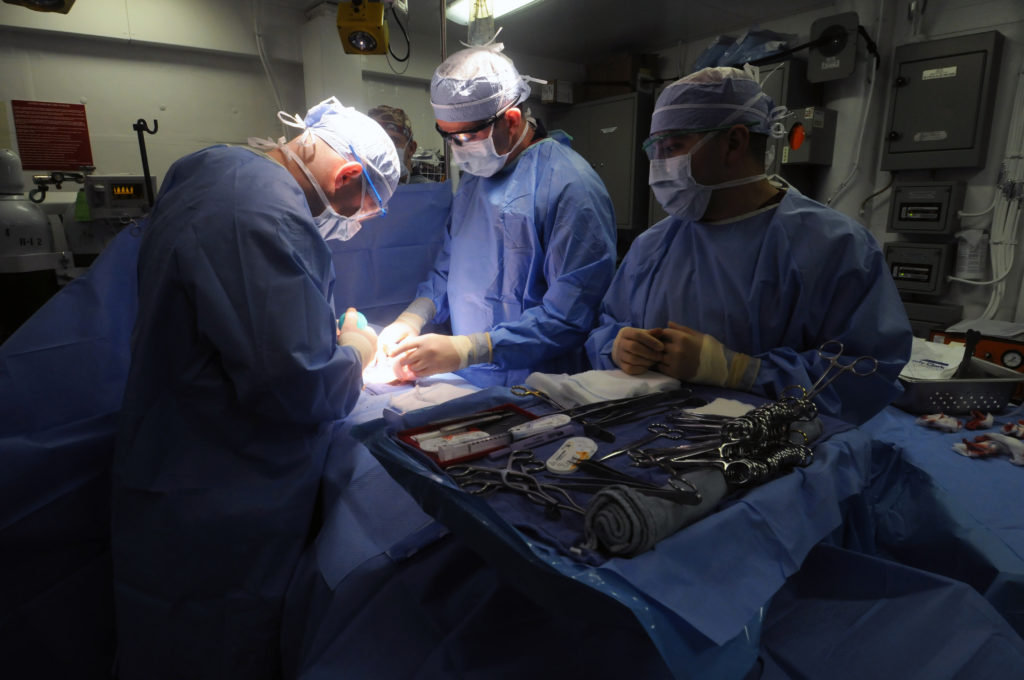
Following lawsuits from thousands of women claiming that transvaginal mesh has inflicted life-altering pain and injury after it was implanted in them, the US Food and Drug Administration (FDA) announced Tuesday a ban on the sale of the surgical device.
Two manufacturers, Boston Scientific and Coloplast, were indicated by the FDA as not having demonstrated a reasonable assurance of safety and effectiveness for these devices.
The surgical mesh is generally used to repair pelvic organ prolapse (POP), but reported side effects include urinary issues, infections and pain during sexual intercourse.
In 2016, the FDA reclassified the mesh as class III or high risk, which meant that manufacturers had to submit applications with the agency and receive approval in order to keep selling the devices in the US.
The agency wanted evidence that showed the mesh worked better than surgery without using the mesh to repair POP, said Jeffrey Shuren, director of the FDA’s Center for Devices and Radiological Health, in a statement.
“That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” he said.
The companies will now have 10 days to submit their plan to withdraw these products from the market, the FDA said.
“Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions,” added Shuren. “The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
Women who have had the procedure should continue with routine check-ups. No additional measures are needed if they are satisfied with their surgery and don’t have complications or symptoms, the FDA said Tuesday.
Those who experience complications or symptoms, such as persistent vaginal bleeding or discharge, pelvic or groin pain or pain with sex, should see their health care provider. While women who were planning to have transvaginal mesh repair of POP should discuss other treatment options with their doctors, the FDA advised.
Boston Scientific said it was deeply disappointed by the agency’s decision, in a statement to Bloomberg.
The company said the inaccessibility of the products will severely limit treatment options for 50 percent of women in the US suffering from pelvic organ prolapse.
“Patient safety is always our highest priority, and we will work closely with the agency to understand its direction and determine next steps,” the company said.
Boston Scientific was the subject of a CBS “60 Minutes” investigation from last May that claimed the manufacturer was facing 48,000 lawsuits related to the device.









Join or login to leave a comment
JOIN LOGIN