Because patients often take more than one medication at a time, there is a risk that drug-drug interactions (DDIs) will result in unanticipated morbidity and mortality. In some cases, DDIs can result in the withdrawal of approved drugs — such as mibefradil, terfenadine, cisapride, and cerivastatin — from the market.
The science of pharmacokinetic DDIs is a relatively new discipline that can reduce the risk of drug-drug interactions. A greater understanding of DDIs, improved testing/modelling capabilities and increasing prevalence of polydrug use have prompted regulators to continually refine and update their recommendations to industry. Those who understand the science and leverage the information derived from in vitro and clinical studies may benefit from expedited drug development timelines and reduced costs including the potential to bypass clinical DDI studies.
The changes to regulatory recommendations can cause confusion for trial sponsors. For instance, sponsors might wonder: what is the most appropriate timing for DDI studies? Which enzymes and transporters should be evaluated? What’s the role of cocktail and microdosing studies in DDI studies? How do we select probe drugs and determine the best study design?
These questions and more are answered in an informative webinar in which Dr. Carol Collins, senior clinical pharmacologist at Medpace, takes a deep dive into each of these topics. She has over 20 years of experience in clinical DDI studies and is a licensed physician with certifications as a principal investigator and regulatory affairs. To learn more about what’s new in clinical DDI studies, watch this on-demand webinar from Medpace, a global clinical contract research organization.
Clinical pharmacokinetic DDI programs have shifted away from empirical approaches — where a sponsor would look at their indication and determine what concomitant medications would likely be prescribed with their investigational drug — to mechanistic approaches — where a sponsor would evaluate an investigational drug’s potential to interact with drug-metabolizing enzymes and transporters. This shift is born out of the knowledge that these enzymes and transporters behave predictably, thus a sponsor can utilize decision trees and mathematical modelling to predict DDIs.
Qualitative & Quantitative Estimations of DDIs
Decision trees give a qualitative ‘yes or no’ answer as to whether a clinical study is required. Through this approach, sponsors need to understand the role, if any, of drug metabolism and transport in drug absorption, distribution and elimination. As sponsors progress through the decision trees, it may be apparent that the drug is not a substrate of drug-metabolizing enzymes or transporters. Or conversely, the investigational drug is potentially an inducer of key enzyme(s) or transporter(s). For the latter, additional clinical studies may be required.
Physiologically based pharmacokinetic (PBPK) modelling can give a qualitative estimation or quantitative estimation of DDI. This type of pharmacokinetic modelling can predict and simulate the magnitude of interactions and evaluate the impact of intrinsic and extrinsic factors.
“A PBPK model can be as simple or as complicated as needed for the situation,” says Collins. “It can be quite a complex modelling situation where you include the physical and chemical properties of the drug, in vitro absorption, distribution, metabolism and elimination (ADME), DDI data and clinical properties from Phase I trials.”
PBPK modelling can be especially helpful for rare disease indications, oncology indications and special patient populations where it would be unsafe for the patients or impractical to conduct the studies.
“For example, it’s very difficult to recruit patients for severe renal or hepatic impairment studies,” says Collins. “Furthermore, there are ethical issues with performing DDI studies in pregnant women and in pediatric [populations]… In these cases, PBPK modeling can actually be used to predict not only the magnitude of an interaction but it can also predict the impact of altering dose regimens without actually conducting a clinical study.”
Collins emphasizes that PBPK modelling needs to be robust and taken very seriously by companies so that regulators can be confident in the assessment.
Timing of Drug-Drug Interaction Studies
The Japanese Ministry of Health, Labour and Welfare (MHLW), European Medicines Agency (EMA) and FDA all say that in vitro studies are generally required before Phase I studies. Furthermore, the latest iteration of the FDA draft guidance states that sponsors should also evaluate the potential for inhibition and induction of key enzymes and transporters before the drug is given to patient populations likely to be taking concomitant medications.
These three regulatory bodies also agree that in vivo studies should be conducted on humans and not animals. That’s largely because there are considerable differences between the drug metabolizing enzymes and transporters between species, and in some instances, the drug causes unexpected adverse events in humans despite passing animal testing.
This issue is clearly described in a 2018 review paper by Dr. Paul Watkins of UNC Eshelman School of Pharmacy at Chapel Hill, North Carolina, who wrote,
“One major reason preclinical models fail to detect intrinsic drug-induced liver injury liability is due to differences in drug metabolism and disposition… ibuprofen, clearly among the safest drugs on the market, can cause kidney and liver failure at therapeutic doses in the dog. If ibuprofen were a new drug addressing an unmet medical need, and the dog was used in preclinical testing, it probably would never progress into Phase I clinical studies.”
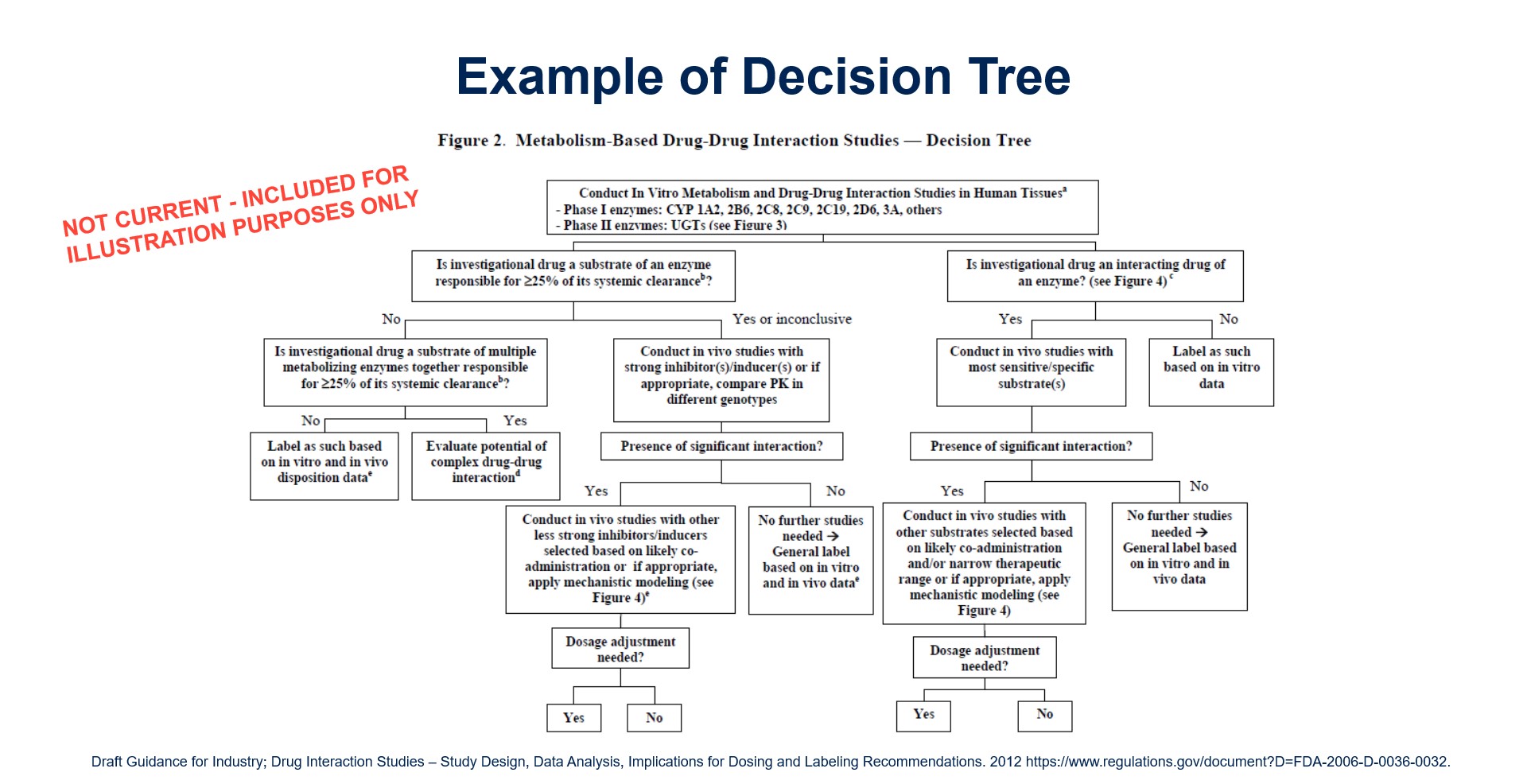
Choosing Enzymes and Transporters for Evaluation
This is an important decision to make prior to beginning a clinical development program. The standard metabolic enzyme panel may include CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4.
In some cases, companies may need to focus on non-P450 enzymes especially if there is evidence that non-P450 enzymatic metabolism — especially phase II enzymes — plays a major role in drug elimination.
Drug transporters may play important roles in drug absorption, distribution and elimination including the concentration at the site(s) of pharmacodynamic action.
The science of drug transport is less mature than our understanding of drug metabolism. This means that regulatory positions are continually evolving in terms of what transporters should be evaluated or the most optimal probe drugs to utilize in those evaluations.
Collins adds, “We recognize that sometimes there are complex metabolic transporter interactions that particularly cause challenges because systemic plasma concentrations may not reflect intracellular concentrations at the site of action. So, we may see pharmacodynamic effects or adverse events that are not in alignment with the plasma concentration [observed].”
The Role of Cocktail and Microdosing Studies
A cocktail study involves concurrent administration of multiple probe substrates to evaluate the inhibition potential for multiple metabolic enzymes and transporters simultaneously. The main advantage of implementing this study design is to improve the efficiency of the study, saving sponsors time and money conducting multiple clinical DDI studies.
These studies are also useful to minimize confounding due to pharmacokinetic variability over time.
“An example of this would be inflammatory diseases that produce cytokines that decrease the expression of key drug metabolites and enzymes,” says Collins.
Sponsors are encouraged to use pre-validated cocktail combinations. However, regulatory agencies may allow the use of new cocktails that have not been clinically validated if the sponsor is working with a challenging patient population such as an orphan disease indication.
Microdosing, as defined by regulatory authorities, involves giving a dose that’s one percent of the pharmacologically active dose. In practice, however, this strategy is sometimes utilized to refer to small dose studies that are low but do not meet this strict definition. The primary reason for using this approach is to minimize potential safety issues. Ideally, drugs should exhibit linear pharmacokinetics, otherwise, it will be impossible to predict the impact of a DDI in the therapeutic range.
Selection of Probe Drugs and Duration of Dosing
The selection of probe drugs continues to evolve. Previously, ketoconazole was the probe drug of choice for CYP3A4 inhibition studies; however safety issues have necessitated the selection of an alternative probe inhibitor. Current industry recommendation for a CYP3A4 inhibitor is itraconazole. Itraconazole exhibits competitive inhibition but it has nonlinear pharmacokinetics and its metabolites contribute to CYP3A4 inhibition.
In contrast, there is no industry consensus on which P-glycoprotein (P-gp) substrate to use.
Digoxin was once the probe substrate of choice, but newer data suggests that digoxin is neither specific or sensitive to P-gp. Other candidates are not sensitive enough to P-gp inhibition or insufficient quantities reach systemic circulation. As a result, there is currently no regulatory or industry consensus on which P-gp substrate to use in clinical DDI studies.
There is general agreement that rifampin is the probe inducer for drug-metabolizing enzymes (CYP2C9, CYP2C19 and CYP3A4/5) and transporters (P-gp) that are regulated by the nuclear receptor PXR. Historically, the EMA has suggested a longer duration of rifampin in clinical DDI studies. Recent publications based on an in-depth evaluation of existing clinical pharmacokinetic data from DDI studies and PBPK modeling support the EMA recommendations.
General Clinical DDI Study Design Principles

There are multiple factors to consider when designing clinical DDI studies. Key differences emerge when the investigational drug is evaluated as a potential substrate (victim) or as a precipitant (perpetrator).
Collins concludes, “I strongly recommend based on the stance of the regulatory authorities that you start your DDI program early and then you carefully assess your program as to whether if there are opportunities to streamline your clinical development program.
Dr. Carol Collins shares more insights on clinical DDI studies in this on-demand webinar.
This article was created in collaboration with the sponsoring company and the Xtalks editorial team.








Join or login to leave a comment
JOIN LOGIN